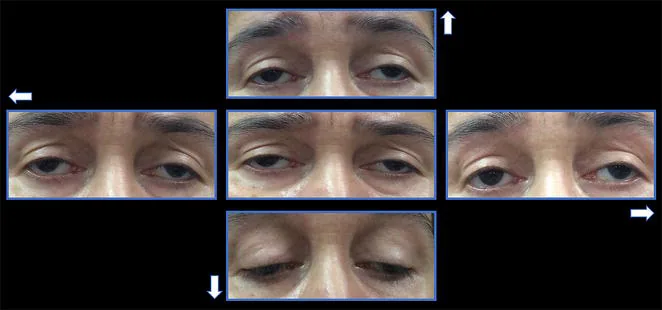
Chronic progressive external ophthalmoplegia (CPEO) is a type of mitochondrial encephalomyopathy, a group of systemic diseases caused by mitochondrial dysfunction. It selectively affects the extraocular muscles, leading to slowly progressive bilateral ptosis and limitation of eye movements.
It is considered the most common phenotype among mitochondrial diseases. According to a UK cohort database, the prevalence is approximately 1/30,000 and the incidence is 1–2/100,000 1).
The history of CPEO dates back to the first description by von Graefe in 1868. In 1958, Kearns and Sayre described the triad of CPEO, retinitis pigmentosa, and cardiac conduction defects. In 1972, ragged red fibers were found on muscle biopsy, and mtDNA deletions were detected in 1988–1989. In 2000, the first nuclear DNA (nDNA) mutation associated with multiple mtDNA deletions was identified 1).
Clinically, CPEO is broadly divided into the following two types.
Isolated CPEO: Ocular symptoms only, without systemic signs. Typical onset is in the 30s to 40s.
CPEO-plus: Accompanied by systemic symptoms such as sensorineural hearing loss, dysphagia, cardiac conduction defects, cerebellar ataxia, and proximal muscle weakness. Kearns-Sayre syndrome (KSS) is a representative example.
Some cases develop around puberty, and due to the lack of systemic clinical signs, patients often first visit an ophthalmologist.
QWhat is the difference between CPEO and CPEO-plus?
A
Isolated CPEO presents only with ptosis and ophthalmoplegia, without systemic symptoms. CPEO-plus is associated with systemic symptoms such as sensorineural hearing loss, dysphagia, cardiac conduction defects, and cerebellar ataxia. Kearns-Sayre syndrome is a representative CPEO-plus disorder that begins before age 20 and includes retinitis pigmentosa and cardiac conduction defects.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
The onset of CPEO symptoms is extremely gradual. The initial symptom is often ptosis.
Ptosis: Patients visit with complaints such as “somehow my eyelids feel heavy.” It gradually progresses from unilateral to bilateral. It is not uncommon for patients to be unaware until pointed out by others.
Ophthalmoplegia: Appears several years after ptosis. Limitation of eye movement in all directions progresses gradually.
Lack of diplopia: Because both eyes are symmetrically affected, patients often do not notice diplopia even with marked ophthalmoplegia.
Compensatory head posture: The patient compensates for limited eye movement by turning the head.
Exercise intolerance: Patients with generalized muscle weakness complain of easy fatigability.
Painlessness is a characteristic of CPEO. Eye pain, proptosis, or pupillary abnormalities suggest other etiologies.
Clinical Findings (Findings Confirmed by Physician Examination)
Ptosis: Bilateral, symmetric, with levator function reduced to less than 8–10 mm 1). In advanced cases, severe ptosis occurs.
Restriction of eye movement: In all directions. Exotropia may be present in primary position.
Pupil sparing: CPEO does not involve pupillary abnormalities. If mydriasis is present, consider other causes such as oculomotor nerve palsy.
Lagophthalmos: Along with ptosis, weakness of the orbicularis oculi muscle leads to incomplete eyelid closure, which may cause exposure keratitis.
Retinal findings: Fluorescein angiography may show granular hyperfluorescence at the level of the retinal pigment epithelium. If salt-and-pepper retinopathy is present, it suggests Kearns-Sayre syndrome.
Because CPEO affects both eyes symmetrically and slowly, there is little difference in eye movement between the two eyes, making diplopia less likely. Also, due to the very slow progression, central adaptation occurs. Only about one-third of patients experience constant or intermittent diplopia.
The causes of CPEO are broadly divided into mitochondrial DNA (mtDNA) mutations and nuclear DNA (nDNA) mutations.
mtDNA Mutations
Single large deletion: The most common cause, accounting for 60–80% of cases. Most deletions range from 1.3 to 1.9 kb. Sporadic cases suggest de novo mutations.
Point mutations: tRNA genes are hot spots. At least 35 point mutations can manifest as isolated CPEO. The MT-TN gene (tRNAAsn) is a hot spot for sporadic PEO, with 7 out of 11 mutations presenting a PEO phenotype 2).
Inheritance pattern: Maternal inheritance. Heteroplasmy (coexistence of normal and mutant mtDNA) is characteristic; when the proportion of mutant mtDNA exceeds 80%, functional impairment becomes apparent at the cellular level2)3).
nDNA mutations
mtDNA maintenance-related genes: Mutations have been reported in POLG (DNA polymerase gamma), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK, and others1)5).
Multiple mtDNA deletions: nDNA mutations cause multiple mtDNA deletions through impaired mtDNA replication and repair.
Inheritance pattern: Autosomal dominant or autosomal recessive inheritance. Taking a family history is important.
The reason extraocular muscles are selectively affected is that they have a higher mitochondrial content and greater metabolic demand compared to skeletal muscle, making them more vulnerable to oxidative phosphorylation impairment.
QIs CPEO hereditary?
A
The inheritance pattern varies depending on the causative gene. Sporadic cases of mtDNA mutations are de novo mutations with low genetic risk. Cases with nDNA mutations show autosomal dominant or recessive inheritance and may occur in families. Even mtDNA mutations can be transmitted through maternal inheritance. Genetic counseling is recommended.
CPEO is primarily a clinical diagnosis, but various tests are useful for confirmation and differential diagnosis. In specialized clinics, attention may be focused on specific diseases, and ptosis or ocular movement disorders may be overlooked 6).
Blood tests: Serum lactate, creatine kinase (CK), and cerebrospinal fluid lactate may be elevated, but sensitivity and specificity are not high. Elevated blood pyruvate and increased lactate/pyruvate ratio are also informative.
Antibody tests: Negative results for anti-acetylcholine receptor antibodies and thyroid autoantibodies are useful for excluding myasthenia gravis and thyroid eye disease.
Biomarkers: Fibroblast growth factor 21 (FGF-21) and growth differentiation factor 15 (GDF-15) are gaining attention as noninvasive screening tools 1).
Electromyography of the extraocular muscles shows interference patterns with sufficient discharge, albeit with slightly weaker amplitude than normal muscles, even when eye movement is not possible. This is useful for differentiating from neurogenic diseases.
mtDNA deletion: Southern blot analysis using skeletal muscle biopsy samples is reliable. Long PCR may allow detection in peripheral blood in some cases.
mtDNA point mutations: Whole mtDNA analysis by next-generation sequencing (NGS) is effective. They may be undetectable in blood and only detected in muscle tissue 2)3).
nDNA mutations: Exome analysis of related genes such as POLG, TWNK, and SLC25A4 is performed 5).
Brain MRI: May show white matter hyperintensities, cortical atrophy, cerebellar atrophy, and brainstem hyperintensities. In one case of KSS, unilateral cerebellar atrophy progressed over time 7).
MR spectroscopy (MRS): Can noninvasively detect lactate accumulation in muscles and may be useful for disease monitoring 4).
Myasthenia gravis is the most important differential diagnosis because it causes ptosis and eye movement disorders. Myasthenia gravis is characterized by diurnal variation and easy fatigability, and improvement is seen with the Tensilon test or ice pack test. Ptosis in CPEO is non-fluctuating and does not improve with these tests.
Indications: When levator function is moderately preserved or better.
Method: Advancement or resection of the levator palpebrae superioris muscle. The aponeurosis is advanced and fixed via a transcutaneous approach, with or without additional tucking of the Müller muscle.
Note: In CPEO, levator function progressively worsens, so the effect may diminish over the long term.
Frontalis Suspension
Indications: When levator function is poor (less than 4 mm). This is often chosen in CPEO.
Method: Connect the upper eyelid to the frontalis muscle using autologous fascia (fascia lata, temporalis fascia), Gore-Tex® sheet, nylon thread, silicone rod, etc.
Prism lenses: Effective for reducing diplopia in small-angle misalignment.
Strabismus surgery: Shortening of the extraocular muscle with impaired movement to correct the primary position. If the angle is large, recession of the antagonist muscle may also be performed. Due to the progressive nature of the disease, recurrence is possible.
Cardiac function evaluation: KSS carries a risk of cardiac conduction defects (AV block), requiring regular ECG and consideration of pacemaker implantation.
Endocrine abnormalities: Management of diabetes, growth hormone deficiency, short stature, etc., is required.
Hearing evaluation: Regular hearing tests are recommended for sensorineural hearing loss.
QCan ptosis recur after surgery?
A
Since CPEO is a progressive disease, levator function may gradually worsen after surgery, leading to recurrence of ptosis. In children, reoperation is often necessary due to growth. The materials used in frontalis suspension may also lose tensile strength over time. Regular follow-up is important.
The essence of CPEO is impairment of oxidative phosphorylation due to mtDNA or nDNA mutations.
Mitochondria have their own DNA (mtDNA), which encodes 13 proteins necessary for oxidative phosphorylation. Deletions or point mutations in mtDNA reduce the activity of electron transport chain enzymes, leading to insufficient ATP production. Extraocular muscles have higher mitochondrial content compared to skeletal muscles and have greater metabolic demands to maintain fatigue resistance. This characteristic is considered one reason why extraocular muscles are selectively affected in CPEO.
Heteroplasmy is an important pathological concept in CPEO. It is a phenomenon where normal and mutant mtDNA coexist within a single cell. When the proportion of mutant mtDNA exceeds a threshold (usually over 80%), the mitochondria become dysfunctional.
Visuttijai et al. (2021) reported two novel point mutations in the MT-TN gene (m.5669G>A and m.5702delA). Single muscle fiber analysis showed that the mutation load in COX-negative fibers averaged 93%, significantly different from COX-normal fibers (32% and 57%) (P < 0.0001). The threshold for COX dysfunction was over 80% in both cases 2).
Katayama Ueda et al. (2025) identified a novel mutation in the tRNAGlu gene (m.14677T>C) in a Japanese male CPEO case. The median mutation load in ragged red fibers was 88.1%, significantly higher than in non-ragged red fibers (median 17.1%) (P = 0.03) 3).
Regarding nDNA mutations, the POLG gene is one of the most common nuclear gene causes of CPEO. POLG encodes DNA polymerase γ, which is necessary for mtDNA replication; its mutations cause multiple mtDNA deletions through impaired mtDNA replication.
Liu et al. (2023) reported a 38-year-old woman with a known mutation c.2857C>T (p.R953C) and a novel mutation c.2391G>C (p.M797I) in the POLG gene. She developed ptosis in addition to limb weakness and numbness, and muscle biopsy revealed ragged red fibers 5).
7. Latest Research and Future Perspectives (Investigational Reports)
In childhood-onset mitochondrial encephalomyopathy, combination therapy with 5-aminolevulinic acid (5-ALA) and iron has been suggested to enhance ATP production, and clinical trials have begun. However, its efficacy in CPEO is unknown.
KH176 is a mitochondrial-targeted redox modulator that reduces reactive oxygen species-mediated cell damage.
A Phase II clinical trial evaluating the safety and efficacy of KH176 (100 mg twice daily) in CPEO patients has been initiated (NCT04604548)5).
Attempts are being made to introduce normal genes using safe vectors. In addition, a method to suppress mutant mtDNA with oligo RNA utilizing the characteristics of heteroplasmy has been reported.
As mitochondrial genome manipulation techniques, selective removal of mutant mtDNA using restriction enzymes, TALEN, ZFN, and CRISPR is being studied1). Replacement therapy in the germline (pronuclear transfer, oocyte spindle transfer) is also at the stage of animal models and clinical trials.
Fan et al. (2021) detected a double peak at 1–2 ppm (suggesting lactate accumulation) in muscle MR spectroscopy of patients with CPEO-plus syndrome. It was more prominent in edematous muscles, indicating potential as a biomarker for disease monitoring4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.