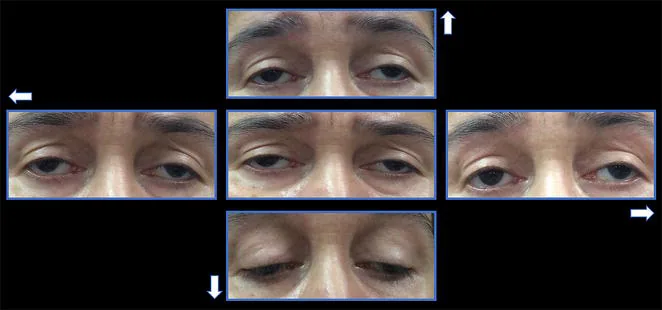
A oftalmoplegia externa progressiva crônica (chronic progressive external ophthalmoplegia: CPEO) é um tipo de doença sistêmica (encefalomiopatia mitocondrial) decorrente de disfunção mitocondrial. Acomete seletivamente os músculos extraoculares, causando ptose bilateral lentamente progressiva e restrição dos movimentos oculares.
É considerada o fenótipo mais comum das doenças mitocondriais. Segundo um banco de dados de coorte do Reino Unido, a prevalência é de aproximadamente 1/30.000 e a incidência de 1-2/100.000 1).
A história da CPEO começa com a primeira descrição por von Graefe em 1868. Em 1958, Kearns e Sayre descreveram a tríade CPEO, retinite pigmentosa e distúrbio de condução cardíaca. Em 1972, fibras vermelhas rasgadas foram descobertas na biópsia muscular, e em 1988-1989 a deleção do mtDNA foi detectada. Em 2000, a primeira mutação no DNA nuclear (nDNA) associada a deleções múltiplas do mtDNA foi identificada 1).
Clinicamente, a CPEO é dividida nos dois tipos a seguir.
CPEO isolada: Apenas sintomas oculares sem sinais sistêmicos. Início típico entre 30 e 40 anos.
CPEO-plus: Acompanhada de sintomas sistêmicos como perda auditiva neurossensorial, disfagia, distúrbios de condução cardíaca, ataxia cerebelar e fraqueza muscular proximal. A síndrome de Kearns-Sayre (KSS) é o exemplo típico.
Alguns casos iniciam antes ou depois da puberdade, com poucos sinais clínicos sistêmicos, frequentemente levando o paciente ao oftalmologista primeiro.
QQual a diferença entre CPEO e CPEO-plus?
A
CPEO isolada envolve apenas ptose e distúrbio da motilidade ocular sem sintomas sistêmicos. CPEO-plus é acompanhada de sintomas sistêmicos como perda auditiva neurossensorial, disfagia, distúrbios de condução cardíaca e ataxia cerebelar. A síndrome de Kearns-Sayre é uma doença CPEO-plus típica que começa antes dos 20 anos, com retinite pigmentosa e distúrbios de condução cardíaca.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
O início dos sintomas da CPEO é muito lento. O primeiro sintoma é frequentemente a ptose.
Ptose: O paciente queixa-se de “dificuldade em levantar a pálpebra”. Progride gradualmente de unilateral para bilateral. Muitas vezes não é percebido até que outra pessoa o note.
Distúrbio da motilidade ocular: Aparece alguns anos após a ptose. A restrição de movimento em todas as direções progride gradualmente.
Ausência de diplopia: Como ambos os olhos são afetados simetricamente, frequentemente não há queixa de diplopia, mesmo com distúrbio significativo da motilidade.
Posição compensatória da cabeça: Compensa a limitação dos movimentos oculares alterando a posição da cabeça.
Intolerância ao exercício: Em casos com fraqueza muscular geral, o paciente queixa-se de fadiga fácil.
A ausência de dor é característica da CPEO. Dor ocular, proptose ou anormalidades pupilares sugerem outras causas.
Achados Clínicos (achados confirmados pelo médico ao exame)
Ptose: Bilateral, simétrica, com função do elevador reduzida para menos de 8-10 mm 1). Em casos avançados, a ptose torna-se grave.
Restrição dos movimentos oculares: Em todas as direções. Pode haver exotropia na posição primária.
Pupilas preservadas: A CPEO não apresenta anormalidades pupilares. Se houver midríase, considere outras causas, como paralisia do nervo oculomotor.
Lagoftalmo: Juntamente com a ptose, ocorre fraqueza do músculo orbicular do olho, resultando em fechamento incompleto das pálpebras, podendo haver ceratite de exposição.
Achados retinianos: A angiografia fluoresceínica pode mostrar hiperfluorescência granular ao nível do epitélio pigmentar da retina. Se acompanhada de retinopatia sal e pimenta, sugere síndrome de Kearns-Sayre.
QPor que os pacientes com CPEO raramente percebem diplopia?
A
Como a CPEO afeta ambos os olhos de forma simétrica e lenta, a diferença nos movimentos oculares é pequena, dificultando a ocorrência de diplopia. Além disso, a progressão é muito lenta, permitindo adaptação central. Apenas cerca de um terço dos pacientes apresenta diplopia persistente ou intermitente.
As causas da CPEO são divididas principalmente em mutações do DNA mitocondrial (mtDNA) e mutações do DNA nuclear (nDNA).
Mutações do mtDNA
Grande deleção única: A causa mais comum, responsável por 60-80% dos casos. A maioria são deleções na faixa de 1,3-1,9 kb. Casos esporádicos sugerem mutação de novo.
Mutações pontuais: Genes de tRNA são pontos críticos. Pelo menos 35 mutações pontuais podem se manifestar como CPEO isolado. O gene MT-TN (tRNAAsn) é um ponto crítico para PEO esporádico, com 7 das 11 mutações apresentando fenótipo PEO 2).
Padrão de herança: Herança materna. Heteroplasmia (mistura de mtDNA normal e mtDNA mutante) é característica, e quando a proporção de mutação excede 80%, a disfunção em nível celular se torna evidente2)3).
Mutações no nDNA
Genes relacionados à manutenção do mtDNA: Mutações foram relatadas em POLG (DNA polimerase γ), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK e outros1)5).
Deleções múltiplas de mtDNA: Mutações no nDNA causam deleções múltiplas de mtDNA através do comprometimento da replicação e reparo do mtDNA.
Padrão de herança: Herança autossômica dominante ou recessiva. A história familiar é importante.
A razão pela qual os músculos extraoculares são seletivamente afetados é que eles têm maior conteúdo mitocondrial e maior demanda metabólica em comparação com os músculos esqueléticos, tornando-os mais vulneráveis ao distúrbio da fosforilação oxidativa.
QCPEO é hereditário?
A
O padrão de herança difere conforme o gene causador. Casos esporádicos de mutações no mtDNA são mutações de novo e o risco hereditário é baixo. Casos de mutações no nDNA mostram herança autossômica dominante ou recessiva, com ocorrência familiar. Mutações no mtDNA também podem ser transmitidas por herança materna. O aconselhamento genético é recomendado.
O CPEO é diagnosticado clinicamente, mas vários exames são úteis para confirmação e diagnóstico diferencial. Em clínicas especializadas, a atenção pode se concentrar em doenças específicas, levando à negligência de ptose ou distúrbios dos movimentos oculares 6).
Exames de sangue: Pode haver aumento do lactato sérico, creatina quinase (CK) e lactato no líquido cefalorraquidiano, mas a sensibilidade e especificidade não são altas. O aumento do piruvato sanguíneo ou da relação lactato/piruvato também é útil.
Testes de anticorpos: A confirmação da negatividade dos anticorpos anti-receptor de acetilcolina e autoanticorpos tireoidianos é útil para excluir miastenia gravis e oftalmopatia tireoidiana.
Biomarcadores: O fator de crescimento de fibroblastos 21 (FGF-21) e o fator de diferenciação de crescimento 15 (GDF-15) têm atraído atenção como triagem não invasiva 1).
Este é o exame chave para o diagnóstico definitivo.
Coloração de Gomori tricrômico: Mostra fibras vermelhas rasgadas (ragged red fibers). As mitocôndrias acumuladas dentro e ao redor das fibras musculares coram-se intensamente.
Dupla coloração COX/SDH: Fibras negativas para citocromo c oxidase (COX) aparecem em um padrão de mosaico.
Microscopia eletrônica: Mostra mitocôndrias gigantes com morfologia anormal entre as fibras musculares.
Na eletromiografia dos músculos extraoculares, observa-se um padrão de interferência com descarga suficiente, embora com amplitude ligeiramente mais fraca que o músculo normal, mesmo quando o movimento ocular não é possível. Isso é útil para diferenciar de doenças neurogênicas.
Deleção de mtDNA: O método de Southern blot usando amostra de biópsia de músculo esquelético é confiável. A PCR longa pode detectar no sangue periférico em alguns casos.
Mutação pontual de mtDNA: A análise completa do mtDNA por sequenciamento de próxima geração (NGS) é eficaz. Às vezes não é detectável no sangue, apenas no tecido muscular 2)3).
Mutações do nDNA: A análise do exoma de genes relacionados, como POLG, TWNK, SLC25A4, é realizada 5).
RM de Crânio: Podem ser observados hipersinal na substância branca, atrofia cortical, atrofia cerebelar e hipersinal no tronco encefálico. Em um caso de KSS, foi relatada atrofia cerebelar unilateral progressiva ao longo do tempo 7).
Espectroscopia de RM (ERM): Pode detectar de forma não invasiva o acúmulo de lactato intramuscular, sendo potencialmente útil para o monitoramento da doença 4).
Atrofia muscular predominante distal. Miotonia de percussão
A miastenia gravis é a doença diferencial mais importante, pois causa ptose e distúrbios dos movimentos oculares. A miastenia gravis é caracterizada por flutuação diurna e fatigabilidade fácil, com melhora no teste de Tensilon e no teste de bolsa de gelo. A ptose na CPEO é não flutuante e não melhora com esses testes.
Indicação: Quando a função do músculo levantador está moderadamente preservada ou melhor.
Método: Realiza-se avanço ou ressecção do músculo levantador da pálpebra superior. Por abordagem percutânea, a aponeurose é avançada e fixada, e, se necessário, adiciona-se plicatura do músculo de Müller.
Nota: Na CPEO, a função do músculo levantador piora progressivamente, portanto o efeito pode diminuir a longo prazo.
Suspensão no Músculo Frontal
Indicações: Quando a função do músculo levantador é ruim (menos de 4 mm). Na CPEO, esta é frequentemente a opção escolhida.
Método: Conectar a pálpebra superior ao músculo frontal usando fáscia autóloga (fáscia lata ou temporal), lâmina de Gore-Tex®, fio de náilon ou haste de silicone.
Atenção: A correção excessiva leva a lagoftalmo e exposição da córnea, aumentando o risco de ceratopatia de exposição e úlcera de córnea.
A avaliação pré-operatória cuidadosa por um cirurgião oculoplástico experiente é essencial. Se o fenômeno de Bell estiver ausente, mantenha uma correção insuficiente.
Lentes prismáticas: Eficazes na redução da diplopia para pequenos desvios oculares.
Cirurgia de estrabismo: Encurtamento do músculo com distúrbio de movimento ocular para correção na posição primária. Se o ângulo for grande, a recessão do músculo antagonista também é realizada. Devido à natureza progressiva da doença, há possibilidade de recorrência.
Avaliação da função cardíaca: Na KSS, há risco de distúrbios de condução cardíaca (bloqueio AV), necessitando de ECG regular e decisão sobre implante de marcapasso.
Anormalidades endócrinas: Manejo de diabetes, deficiência de hormônio do crescimento e baixa estatura é necessário.
Avaliação auditiva: Exames auditivos regulares são recomendados para perda auditiva neurossensorial.
QA ptose pode recorrer após a cirurgia?
A
Como a CPEO é uma doença progressiva, a função do músculo levantador pode piorar gradualmente após a cirurgia, levando à recorrência da ptose. Especialmente em crianças, a re-operação é frequentemente necessária com o crescimento. O material usado na cirurgia de suspensão frontal pode perder força de tração a longo prazo. O acompanhamento regular é importante.
A essência da CPEO é um distúrbio da fosforilação oxidativa devido a mutações no mtDNA ou nDNA.
As mitocôndrias possuem seu próprio DNA (mtDNA), que codifica 13 proteínas necessárias para a fosforilação oxidativa. Deleções ou mutações pontuais no mtDNA reduzem a atividade das enzimas da cadeia de transporte de elétrons, causando deficiência na produção de ATP. Os músculos extraoculares têm maior conteúdo mitocondrial em comparação com os músculos esqueléticos e maiores demandas metabólicas para manter a resistência à fadiga. Essa característica é considerada uma das razões pelas quais os músculos extraoculares são seletivamente afetados na CPEO.
A heteroplasmia é um conceito patológico importante na CPEO. É o fenômeno em que mtDNA normal e mtDNA mutante coexistem em uma mesma célula. Quando a proporção de mtDNA mutante excede um limiar (geralmente >80%), essa mitocôndria torna-se disfuncional.
Visuttijai et al. (2021) relataram duas novas mutações pontuais no gene MT-TN (m.5669G>A e m.5702delA). A análise de fibra muscular única mostrou que a proporção média de mutação nas fibras COX-negativas foi de 93%, significativamente diferente das fibras COX-normais (32% e 57%) (P < 0,0001). O limiar de disfunção da COX em ambos os casos foi >80%2).
Katayama Ueda et al. (2025) identificaram uma nova mutação (m.14677T>C) no gene tRNAGlu em um caso de CPEO em um homem japonês. A mediana da proporção de mutação nas fibras ragged red foi de 88,1%, significativamente maior do que nas fibras não ragged red (mediana 17,1%) (P = 0,03)3).
Em relação às mutações no nDNA, o gene POLG é uma das causas nucleares mais comuns de CPEO. O POLG codifica a DNA polimerase γ necessária para a replicação do mtDNA, e suas mutações causam deleções múltiplas do mtDNA através do comprometimento da replicação do mtDNA.
Liu et al. (2023) relataram uma mulher de 38 anos com uma mutação conhecida c.2857C>T (p.R953C) e uma nova mutação c.2391G>C (p.M797I) no gene POLG. Ptose apareceu além de fraqueza e dormência nos membros, e a biópsia muscular confirmou fibras ragged red5).
7. Pesquisas Recentes e Perspectivas Futuras (Relatos em Fase de Pesquisa)
Na encefalomiopatia mitocondrial de início na infância, a terapia combinada de ácido 5-aminolevulínico (5-ALA) e suplementação de ferro foi sugerida para aumentar a produção de ATP, e ensaios clínicos foram iniciados. No entanto, sua eficácia na CPEO ainda é desconhecida.
KH176 é um agente regulador redox direcionado à mitocôndria, reduzindo o dano celular mediado por espécies reativas de oxigênio.
Um ensaio clínico de Fase II para avaliar a segurança e eficácia de KH176 (100 mg duas vezes ao dia) em pacientes com CPEO foi iniciado (NCT04604548)5).
Tentativas estão sendo feitas para introduzir o gene normal usando um vetor seguro. Também foi relatado um método para suprimir o mtDNA mutante com oligo RNA, aproveitando a característica da heteroplasmia.
Como técnicas de manipulação do genoma mitocondrial, a remoção seletiva de mtDNA mutante usando enzimas de restrição, TALEN, ZFN e CRISPR está sendo estudada1). A terapia de substituição na linhagem germinativa (transferência de pronúcleo/transferência de fuso oocitário) também está em estágio de modelos animais e ensaios clínicos.
Fan et al. (2021) detectaram um pico duplo em 1-2 ppm (sugerindo acúmulo de lactato) na espectroscopia de RM muscular em pacientes com síndrome CPEO-plus. Foi mais proeminente em músculos edemaciados, indicando potencial como biomarcador para monitoramento da doença4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.