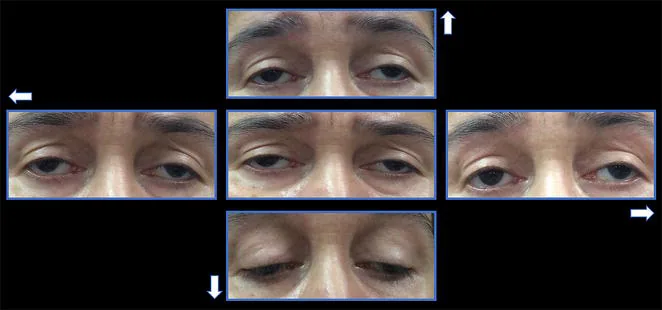
Хроническая прогрессирующая наружная офтальмоплегия (ХПНО) — это форма митохондриальной энцефаломиопатии, группы системных заболеваний, обусловленных митохондриальной дисфункцией. Она избирательно поражает наружные глазные мышцы, вызывая медленно прогрессирующий двусторонний птоз и ограничение подвижности глаз.
Это наиболее частый фенотип митохондриальных заболеваний. По данным британской когортной базы данных, распространенность составляет около 1/30 000, а заболеваемость — 1–2/100 0001).
История ХПНО начинается с первого описания фон Грефе в 1868 году. В 1958 году Кернс и Сейр описали триаду: ХПНО, пигментный ретинит и нарушение проводимости сердца. В 1972 году при мышечной биопсии были обнаружены рваные красные волокна, а в 1988–1989 годах выявлены делеции мтДНК. В 2000 году была идентифицирована первая мутация ядерной ДНК (яДНК), ассоциированная с множественными делециями мтДНК1).
Клинически ХПНО подразделяется на два основных типа.
Изолированная CPEO: только глазные симптомы, без системных проявлений. Типичное начало в возрасте 30–40 лет.
CPEO-plus: сопровождается системными симптомами, такими как нейросенсорная тугоухость, дисфагия, нарушения сердечной проводимости, мозжечковая атаксия, проксимальная мышечная слабость. Синдром Кернса-Сейра (KSS) является типичным примером.
Также встречаются случаи с началом в период полового созревания, скудными системными клиническими признаками, и пациенты часто сначала обращаются к офтальмологу.
QВ чем разница между CPEO и CPEO-plus?
A
Изолированная CPEO проявляется только птозом и нарушением движений глаз без системных симптомов. CPEO-plus включает системные симптомы, такие как нейросенсорная тугоухость, дисфагия, нарушения сердечной проводимости, мозжечковая атаксия. Синдром Кернса-Сейра является типичным заболеванием CPEO-plus, начинающимся до 20 лет, с пигментным ретинитом и нарушениями сердечной проводимости.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
Симптомы CPEO развиваются крайне медленно. Первым симптомом часто является птоз.
Птоз: пациент обращается с жалобой, что «веко как-то плохо поднимается». Постепенно прогрессирует от одностороннего до двустороннего. Нередко пациент не замечает этого, пока кто-то другой не укажет.
Нарушение движений глаз: появляется через несколько лет после птоза. Постепенно прогрессирует ограничение движений во всех направлениях.
Отсутствие диплопии: поскольку оба глаза поражаются симметрично, пациенты часто не ощущают двоения, несмотря на выраженные нарушения движений глаз.
Компенсаторное положение головы: пациент компенсирует недостаток движений глаз изменением положения головы.
Непереносимость физической нагрузки: в случаях с генерализованной мышечной слабостью пациент жалуется на быструю утомляемость.
Отсутствие боли является характерным признаком CPEO. Боль в глазах, экзофтальм или аномалии зрачка указывают на другие причины.
Клинические признаки (обнаруживаемые врачом при осмотре)
Птоз : двусторонний, симметричный, функция леватора снижена до менее 8-10 мм 1). В запущенных случаях выраженный птоз.
Ограничение движений глаз : во всех направлениях. В первичном положении может наблюдаться экзотропия.
Сохранность зрачка : CPEO не сопровождается аномалиями зрачка. Расширение зрачка указывает на другую причину, например, паралич глазодвигательного нерва.
Неполное смыкание век : наряду с птозом из-за слабости круговой мышцы глаза, что может привести к кератиту из-за лагофтальма.
Изменения сетчатки : при флюоресцентной ангиографии может наблюдаться зернистая гиперфлюоресценция на уровне пигментного эпителия сетчатки. Ретинопатия типа «соль с перцем» указывает на синдром Кернса-Сейра.
МРТ орбиты : атрофия наружных глазных мышц.
QПочему при CPEO редко ощущается двоение?
A
При CPEO оба глаза поражаются симметрично и медленно, поэтому разница в движениях глаз невелика, и двоение возникает редко. Кроме того, очень медленное прогрессирование приводит к центральной адаптации. Только примерно у трети пациентов наблюдается постоянное или периодическое двоение.
Причины CPEO делятся на мутации митохондриальной ДНК (мтДНК) и мутации ядерной ДНК (яДНК).
Мутации мтДНК
Одиночная крупная делеция : наиболее частая причина, составляющая 60-80% случаев. Большинство делеций имеют размер 1,3-1,9 т.п.н. Спорадические случаи указывают на мутацию de novo.
Точечные мутации : гены тРНК являются горячими точками. По меньшей мере 35 точечных мутаций могут проявляться как изолированная CPEO. Ген MT-TN (тРНКAsn) является горячей точкой для спорадической PEO, и 7 из 11 мутаций дают фенотип PEO 2).
Тип наследования : материнское наследование. Характерен гетероплазмия (смесь нормальной и мутантной мтДНК); когда доля мутации превышает 80%, на клеточном уровне проявляется дисфункция2)3).
Мутация яДНК
Гены, связанные с поддержанием мтДНК : сообщалось о мутациях в POLG (ДНК-полимераза γ), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK и других1)5).
Множественные делеции мтДНК : мутации яДНК вызывают множественные делеции мтДНК через нарушение репликации и репарации мтДНК.
Тип наследования : аутосомно-доминантный или аутосомно-рецессивный. Важен сбор семейного анамнеза.
Причина избирательного поражения наружных глазных мышц заключается в том, что по сравнению со скелетными мышцами они имеют более высокое содержание митохондрий и большую метаболическую потребность, что делает их более уязвимыми к нарушениям окислительного фосфорилирования.
QПередается ли CPEO по наследству?
A
Тип наследования зависит от гена-причины. Спорадические случаи мутации мтДНК являются мутациями de novo, и генетический риск низок. Случаи мутации яДНК демонстрируют аутосомно-доминантное или рецессивное наследование с семейными случаями. Мутации мтДНК также могут передаваться по материнской линии. Рекомендуется генетическое консультирование.
CPEO в основном диагностируется клинически, но различные исследования полезны для подтверждения диагноза и дифференциальной диагностики. В специализированных клиниках внимание может быть сосредоточено на конкретном заболевании, и птоз или нарушения движений глаз могут быть пропущены 6).
Анализы крови : Может наблюдаться повышение уровня лактата в сыворотке, креатинкиназы (КК) и лактата в спинномозговой жидкости, но чувствительность и специфичность невысоки. Повышение уровня пирувата в крови и соотношения лактат/пируват также может быть полезным.
Анализы на антитела : Подтверждение отрицательного результата антител к ацетилхолиновым рецепторам и аутоантител к щитовидной железе полезно для исключения миастении гравис и эндокринной офтальмопатии.
Биомаркеры : Фактор роста фибробластов 21 (FGF-21) и фактор дифференцировки роста 15 (GDF-15) привлекают внимание в качестве неинвазивного скрининга 1).
Это ключевое исследование для окончательного диагноза.
Окраска по Гомори трихромом : Обнаруживаются рваные красные волокна (ragged red fibers). Накопленные в мышечных волокнах или вокруг них митохондрии интенсивно окрашиваются.
Двойное окрашивание COX/SDH : Волокна, отрицательные по цитохром-с-оксидазе (COX), появляются в виде мозаики.
Электронная микроскопия : Между мышечными волокнами наблюдаются гигантские митохондрии с аномальной морфологией.
Электромиография наружных глазных мышц показывает интерференционную картину с несколько меньшей амплитудой, чем у нормальных мышц, но с достаточной разрядкой даже при отсутствии движений глаз. Полезна для дифференциации с нейрогенными заболеваниями.
Делеция мтДНК : Метод саузерн-блоттинга на образце биопсии скелетных мышц надежен. С помощью метода длинной ПЦР иногда можно обнаружить и в периферической крови.
Точечная мутация мтДНК : Полный анализ мтДНК с помощью секвенирования нового поколения (NGS) эффективен. Иногда она обнаруживается только в мышечной ткани, а не в крови 2)3).
Мутация яДНК: проводится экзомный анализ связанных генов, таких как POLG, TWNK, SLC25A4 5).
МРТ головного мозга: может выявлять гиперинтенсивность белого вещества, корковую атрофию, атрофию мозжечка, гиперинтенсивность ствола мозга и др. В одном случае KSS сообщалось о прогрессирующей односторонней атрофии мозжечка с течением времени 7).
МР-спектроскопия (MRS): может неинвазивно обнаруживать накопление лактата в мышцах, потенциально полезна для мониторинга заболевания 4).
Атрофия мышц с преобладанием дистальных отделов. Перкуссионная миотония
Миастения гравис является наиболее важным дифференциальным диагнозом, поскольку она вызывает птоз и нарушения движений глаз. Для миастении гравис характерны суточные колебания и утомляемость, а также улучшение при проведении тензилонового теста или теста с пакетом льда. Птоз при CPEO не является флуктуирующим и не улучшается при этих тестах.
Показания : при сохранной функции леватора средней или высокой степени.
Метод : выполняется резекция или пликация мышцы, поднимающей верхнее веко. Чрескожным доступом апоневроз перемещается и фиксируется, при необходимости добавляется подшивание мышцы Мюллера.
Примечание : при CPEO функция леватора прогрессивно ухудшается, поэтому эффект может со временем ослабевать.
Подвешивание к лобной мышце
Показания: при плохой функции леватора (менее 4 мм). При ХПНГ часто выбирают этот метод.
Метод: соединение верхнего века с лобной мышцей с использованием аутологичной фасции (фасция бедра, височная фасция), листа Gore-Tex®, нейлоновой нити, силиконового стержня и т.д.
Внимание: гиперкоррекция может привести к лагофтальму и экспозиции роговицы с риском экспозиционной кератопатии и язвы роговицы.
Тщательная предоперационная оценка опытным окулопластическим хирургом обязательна. При отсутствии феномена Белла следует стремиться к гипокоррекции.
Призматические линзы: эффективны для уменьшения диплопии при небольших углах отклонения глаз.
Хирургия косоглазия: укорочение мышцы с нарушением подвижности для коррекции в первичной позиции. При большом угле также выполняется рецессия антагониста. Из-за прогрессирующего характера заболевания возможен рецидив.
Оценка сердечной функции: при KSS существует риск нарушений проводимости сердца (АВ-блокада); необходимы регулярная ЭКГ и решение об установке кардиостимулятора.
Эндокринные нарушения: требуется контроль диабета, дефицита гормона роста, низкорослости и т.д.
Оценка слуха: рекомендуются регулярные аудиометрические исследования при нейросенсорной тугоухости.
QМожет ли птоз рецидивировать после операции?
A
ХПНГ является прогрессирующим заболеванием, поэтому после операции функция леватора может постепенно ухудшаться, что приводит к рецидиву птоза. Особенно у детей с ростом часто требуется повторная операция. Материалы, используемые для фронтальной подвески, также могут со временем терять тяговую силу. Важно регулярное наблюдение.
Сущность CPEO заключается в нарушении окислительного фосфорилирования вследствие мутаций мтДНК или яДНК.
Митохондрии имеют собственную ДНК (мтДНК), которая кодирует 13 белков, необходимых для окислительного фосфорилирования. Делеции или точковые мутации мтДНК снижают активность ферментов дыхательной цепи, что приводит к дефициту АТФ. Наружные глазные мышцы имеют более высокое содержание митохондрий по сравнению со скелетными мышцами и имеют более высокие метаболические потребности для поддержания устойчивости к утомлению. Эта особенность считается одной из причин избирательного поражения наружных глазных мышц при CPEO.
Гетероплазмия является важной патофизиологической концепцией CPEO. Это явление сосуществования нормальной и мутантной мтДНК в одной клетке. Когда доля мутантной мтДНК превышает порог (обычно >80%), митохондрия становится дисфункциональной.
Visuttijai и соавт. (2021) сообщили о двух новых точковых мутациях в гене MT-TN (m.5669G>A и m.5702delA). Анализ одиночных мышечных волокон показал, что средний процент мутации в COX-отрицательных волокнах составил 93%, что значимо отличалось от COX-нормальных волокон (32% и 57%) (P < 0,0001). Порог дисфункции COX в обоих случаях превышал 80% 2).
Katayama Ueda и соавт. (2025) идентифицировали новую мутацию в гене tRNAGlu (m.14677T>C) у японского мужчины с CPEO. Медианный процент мутации в ragged-red волокнах составил 88,1%, что было значимо выше, чем в не-ragged-red волокнах (медиана 17,1%) (P = 0,03) 3).
Что касается мутаций яДНК, ген POLG является одной из наиболее распространенных ядерных генетических причин CPEO. POLG кодирует ДНК-полимеразу γ, необходимую для репликации мтДНК, и его мутации приводят к множественным делециям мтДНК через нарушение репликации.
Liu и соавт. (2023) сообщили о 38-летней женщине с известной мутацией c.2857C>T (p.R953C) и новой мутацией c.2391G>C (p.M797I) в гене POLG. У нее развились слабость и онемение конечностей, а также птоз, и биопсия мышцы выявила ragged-red волокна 5).
7. Новейшие исследования и перспективы на будущее (отчеты исследовательской стадии)
При митохондриальной энцефаломиопатии с началом в детском возрасте комбинированная терапия 5-аминолевулиновой кислотой (5-АЛК) и препаратами железа, как предполагается, повышает продукцию АТФ, и начаты клинические испытания. Однако эффективность при CPEO неизвестна.
KH176 является митохондриально-направленным регулятором окислительно-восстановительного баланса, который уменьшает повреждение клеток, опосредованное активными формами кислорода.
Начато клиническое исследование фазы II по оценке безопасности и эффективности KH176 (100 мг два раза в день) у пациентов с ХПНГ (NCT04604548)5).
Предпринимаются попытки ввести нормальный ген с помощью безопасного вектора. Также сообщается о методе подавления мутантной мтДНК с помощью олиго-РНК, использующем особенности гетероплазмии.
В качестве методов манипуляции с митохондриальным геномом исследуется селективное удаление мутантной мтДНК с помощью рестриктаз, TALEN, ZFN и CRISPR1). Заместительная терапия на уровне зародышевой линии (перенос пронуклеусов, перенос веретена ооцита) также находится на стадии животных моделей и клинических испытаний.
Fan и соавт. (2021) обнаружили двойной пик при 1-2 ppm (указывающий на накопление лактата) при МР-спектроскопии мышц у пациентов с синдромом ХПНГ-плюс. Этот пик был более выражен в отечных мышцах, что указывает на его потенциал в качестве биомаркера для мониторинга заболевания4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.