Die chronisch progressive externe Ophthalmoplegie (CPEO) ist eine Form der mitochondrialen Enzephalomyopathie, einer Gruppe systemischer Erkrankungen aufgrund mitochondrialer Dysfunktion. Sie befällt selektiv die äußeren Augenmuskeln und führt zu einer langsam fortschreitenden beidseitigen Ptosis und Bewegungseinschränkung der Augen.
Sie ist der häufigste Phänotyp mitochondrialer Erkrankungen. Laut einer britischen Kohortendatenbank beträgt die Prävalenz etwa 1/30.000 und die Inzidenz 1–2/100.0001).
Die Geschichte der CPEO beginnt mit der ersten Beschreibung durch von Graefe im Jahr 1868. 1958 beschrieben Kearns und Sayre die Trias aus CPEO, Retinitis pigmentosa und Herzleitungsstörungen. 1972 wurden in der Muskelbiopsie Ragged-Red-Fasern entdeckt, und 1988–1989 wurden mtDNA-Deletionen nachgewiesen. Im Jahr 2000 wurde die erste nukleäre DNA (nDNA)-Mutation identifiziert, die mit multiplen mtDNA-Deletionen einhergeht1).
Klinisch wird die CPEO in zwei Haupttypen unterteilt.
Isoliertes CPEO: Nur Augensymptome, keine systemischen Anzeichen. Typischerweise Beginn in den 30ern bis 40ern.
CPEO-plus: Begleitet von systemischen Symptomen wie Schallempfindungsschwerhörigkeit, Dysphagie, Herzleitungsstörungen, Kleinhirnataxie, proximaler Muskelschwäche. Das Kearns-Sayre-Syndrom (KSS) ist ein repräsentatives Beispiel.
Es gibt auch Fälle mit Beginn um die Pubertät, mit wenigen systemischen klinischen Zeichen, und die Patienten konsultieren oft zuerst einen Augenarzt.
QWas ist der Unterschied zwischen CPEO und CPEO-plus?
A
Isoliertes CPEO zeigt nur Ptosis und Augenbewegungsstörungen ohne systemische Symptome. CPEO-plus umfasst systemische Symptome wie Schallempfindungsschwerhörigkeit, Dysphagie, Herzleitungsstörungen, Kleinhirnataxie. Das Kearns-Sayre-Syndrom ist eine repräsentative Erkrankung des CPEO-plus, die vor dem 20. Lebensjahr beginnt und mit Retinitis pigmentosa und Herzleitungsstörungen einhergeht.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
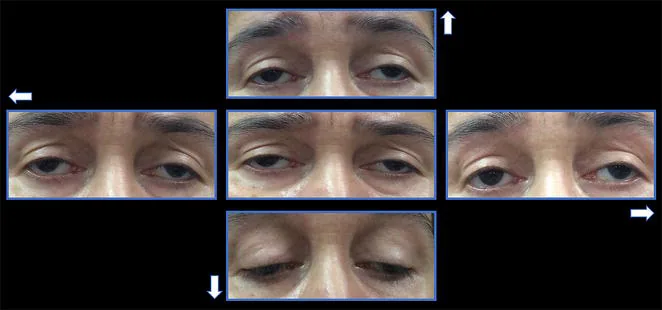
Die Symptome des CPEO treten äußerst langsam auf. Das erste Symptom ist oft eine Ptosis.
Ptosis: Der Patient kommt mit der Beschwerde, dass „das Lid irgendwie nicht gut hochgeht“. Es schreitet allmählich von einseitig zu beidseitig fort. Es ist nicht selten, dass der Patient es erst bemerkt, wenn ein Dritter darauf hinweist.
Augenbewegungsstörungen: Treten einige Jahre nach der Ptosis auf. Eine Bewegungseinschränkung in alle Richtungen schreitet allmählich fort.
Fehlen von Doppelbildern: Da beide Augen symmetrisch betroffen sind, verspüren die Patienten trotz deutlicher Augenbewegungsstörungen oft keine Doppelbilder.
Kompensatorische Kopfhaltung: Der Patient gleicht die unzureichende Augenbewegung durch Veränderung der Kopfhaltung aus.
Belastungsintoleranz: In Fällen mit generalisierter Muskelschwäche klagt der Patient über leichte Ermüdbarkeit.
Schmerzfreiheit ist charakteristisch für CPEO. Augenschmerzen, Exophthalmus oder Pupillenanomalien deuten auf andere Ursachen hin.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellt)
Ptosis : beidseitig, symmetrisch, mit einer Levatorfunktion von weniger als 8-10 mm 1). In fortgeschrittenen Fällen schwere Ptosis.
Einschränkung der Augenbewegungen : in alle Richtungen. In Primärposition kann ein Exotropie vorliegen.
Pupillenerhalt : CPEO geht nicht mit Pupillenanomalien einher. Bei Pupillenerweiterung ist an andere Ursachen wie eine Okulomotoriusparese zu denken.
Lidschlussunfähigkeit : zusammen mit Ptosis aufgrund einer Schwäche des Musculus orbicularis, die zu einer Keratitis e lagophthalmo führen kann.
Netzhautbefunde : In der Fluoreszenzangiographie kann eine granuläre Hyperfluoreszenz auf Höhe des retinalen Pigmentepithels auftreten. Eine Salt-and-Pepper-Retinopathie deutet auf ein Kearns-Sayre-Syndrom hin.
Orbitale MRT : Atrophie der äußeren Augenmuskeln.
QWarum wird bei CPEO selten Doppeltsehen wahrgenommen?
A
Bei CPEO sind beide Augen symmetrisch und langsam betroffen, sodass der Bewegungsunterschied gering ist und Doppeltsehen selten auftritt. Zudem führt die sehr langsame Progression zu einer zentralen Adaptation. Nur etwa ein Drittel der Patienten hat ständiges oder intermittierendes Doppeltsehen.
Die Ursachen der CPEO werden in Mutationen der mitochondrialen DNA (mtDNA) und der nukleären DNA (nDNA) unterteilt.
mtDNA-Mutationen
Große Einzeldeletion : häufigste Ursache mit 60-80 % der Fälle. Die meisten Deletionen sind 1,3-1,9 kb groß. Sporadische Fälle deuten auf eine De-novo-Mutation hin.
Punktmutationen : tRNA-Gene sind Hotspots. Mindestens 35 Punktmutationen können als isolierte CPEO auftreten. Das MT-TN-Gen (tRNAAsn) ist ein Hotspot für sporadische PEO, und 7 von 11 Mutationen zeigen einen PEO-Phänotyp 2).
Vererbungsmodus : mütterliche Vererbung. Heteroplasmie (Mischung aus normaler und mutierter mtDNA) ist charakteristisch; wenn der Mutationsanteil 80 % übersteigt, wird eine Funktionsstörung auf zellulärer Ebene manifest2)3).
nDNA-Mutation
mtDNA-Erhaltungsgene : Mutationen in POLG (DNA-Polymerase γ), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK und anderen wurden berichtet1)5).
Multiple mtDNA-Deletionen : nDNA-Mutationen führen über eine Beeinträchtigung der mtDNA-Replikation und -Reparatur zu multiplen mtDNA-Deletionen.
Vererbungsmodus : autosomal-dominant oder autosomal-rezessiv. Die Erhebung der Familienanamnese ist wichtig.
Der Grund für die selektive Schädigung der äußeren Augenmuskeln ist, dass sie im Vergleich zur Skelettmuskulatur einen höheren Mitochondriengehalt und einen größeren Stoffwechselbedarf haben, was sie anfälliger für Störungen der oxidativen Phosphorylierung macht.
QIst CPEO erblich?
A
Der Vererbungsmodus variiert je nach ursächlichem Gen. Sporadische Fälle von mtDNA-Mutationen sind De-novo-Mutationen mit geringem genetischen Risiko. Fälle mit nDNA-Mutationen zeigen einen autosomal-dominanten oder -rezessiven Erbgang mit familiärem Auftreten. Auch mtDNA-Mutationen können mütterlich vererbt werden. Eine genetische Beratung wird empfohlen.
Die CPEO basiert hauptsächlich auf der klinischen Diagnose, aber verschiedene Untersuchungen sind für die Bestätigung und Differenzialdiagnose nützlich. In Fachkliniken kann die Aufmerksamkeit auf eine bestimmte Erkrankung konzentriert sein, und eine Ptosis oder Augenbewegungsstörungen können übersehen werden 6).
Blutuntersuchungen : Ein Anstieg des Serumlaktats, der Kreatinkinase (CK) und des Liquorlaktats kann auftreten, aber Sensitivität und Spezifität sind nicht hoch. Ein Anstieg des Blutpyruvats und des Laktat/Pyruvat-Quotienten kann ebenfalls hilfreich sein.
Antikörpertests : Der Nachweis negativer Anti-Acetylcholin-Rezeptor-Antikörper und Schilddrüsen-Autoantikörper ist nützlich, um Myasthenia gravis und eine endokrine Orbitopathie auszuschließen.
Biomarker : Fibroblasten-Wachstumsfaktor 21 (FGF-21) und Wachstumsdifferenzierungsfaktor 15 (GDF-15) gewinnen als nicht-invasive Screening-Methoden an Bedeutung 1).
Dies ist die entscheidende Untersuchung für die definitive Diagnose.
Gomori-Trichrom-Färbung : Es zeigen sich Ragged Red Fibers (zerfaserte rote Fasern). Die in oder um die Muskelfasern angesammelten Mitochondrien werden stark angefärbt.
COX/SDH-Doppelfärbung : Cytochrom-c-Oxidase (COX)-negative Fasern treten mosaikartig auf.
Elektronenmikroskopie : Riesige, morphologisch abnorme Mitochondrien werden zwischen den Muskelfasern beobachtet.
Die Elektromyographie der äußeren Augenmuskeln zeigt ein Interferenzmuster mit etwas geringerer Amplitude als bei normalen Muskeln, aber mit ausreichender Entladung, selbst wenn keine Augenbewegungen möglich sind. Nützlich zur Abgrenzung von neurogenen Erkrankungen.
mtDNA-Deletion : Die Southern-Blot-Methode an einer Skelettmuskelbiopsieprobe ist zuverlässig. Mit der Long-PCR-Methode kann sie manchmal auch im peripheren Blut nachgewiesen werden.
mtDNA-Punktmutation : Die vollständige mtDNA-Analyse mittels Next-Generation-Sequencing (NGS) ist effektiv. Manchmal ist sie nur im Muskelgewebe und nicht im Blut nachweisbar 2)3).
nDNA-Mutation: Exom-Sequenzierung verwandter Gene wie POLG, TWNK, SLC25A4 wird durchgeführt 5).
Orbita-MRT: Darstellung der Atrophie der äußeren Augenmuskeln.
Gehirn-MRT: Kann Hyperintensitäten der weißen Substanz, kortikale Atrophie, Kleinhirnatrophie, Hirnstammhyperintensitäten usw. zeigen. Ein Fall von KSS berichtete über eine im Laufe der Zeit fortschreitende einseitige Kleinhirnatrophie 7).
MR-Spektroskopie (MRS): Kann nicht-invasiv die Laktatanreicherung in Muskeln nachweisen, möglicherweise nützlich für die Krankheitsüberwachung 4).
Hypertrophie der äußeren Augenmuskeln. Begleitet von Exophthalmus
Myotone Dystrophie
Muskelatrophie mit Betonung der distalen Muskulatur. Perkussionsmyotonie
Die Myasthenia gravis ist die wichtigste Differentialdiagnose, da sie zu Ptosis und Augenbewegungsstörungen führt. Bei Myasthenia gravis sind Tagesschwankungen und Ermüdbarkeit charakteristisch, und eine Besserung zeigt sich im Tensilon-Test oder Eispack-Test. Die Ptosis bei CPEO ist nicht fluktuierend und bessert sich nicht unter diesen Tests.
Indikation : Bei erhaltener Levatorfunktion mittleren bis guten Grades.
Methode : Vorlagerung oder Resektion des Musculus levator palpebrae superioris. Über einen perkutanen Zugang wird die Aponeurose vorgelagert und fixiert, ggf. mit Tucking des Müller-Muskels.
Hinweis : Bei CPEO verschlechtert sich die Levatorfunktion progredient, sodass der Effekt langfristig nachlassen kann.
Stirnmuskelaufhängung
Indikationen: Bei schlechter Levatorfunktion (weniger als 4 mm). Bei CPEO wird dies oft gewählt.
Methode: Verbindung des Oberlids mit dem Stirnmuskel mittels autologer Faszie (Fascia lata, Temporalfaszie), Gore-Tex®-Folie, Nylonfaden, Silikonstab usw.
Eine sorgfältige präoperative Beurteilung durch einen erfahrenen okuloplastischen Chirurgen ist unerlässlich. Fehlt das Bell-Phänomen, sollte eine Unterkorrektur angestrebt werden.
Prismenlinsen: Wirksam zur Reduktion von Doppelbildern bei kleinem Schielwinkel.
Schieloperation: Verkürzung des beeinträchtigten Augenmuskels zur Korrektur in Primärposition. Bei großem Winkel wird auch eine Rücklagerung des Antagonisten durchgeführt. Aufgrund der fortschreitenden Erkrankung ist ein Rezidiv möglich.
Herzfunktionsbeurteilung: Bei KSS besteht ein Risiko für kardiale Reizleitungsstörungen (AV-Block); regelmäßige EKG-Untersuchungen und die Entscheidung zur Schrittmacherimplantation sind erforderlich.
Endokrine Störungen: Management von Diabetes, Wachstumshormonmangel, Kleinwuchs usw. ist erforderlich.
Hörbeurteilung: Regelmäßige Hörtests bei Schallempfindungsschwerhörigkeit werden empfohlen.
QKann ein Ptosis nach einer Operation wieder auftreten?
A
Da CPEO eine fortschreitende Erkrankung ist, kann die Levatorfunktion auch nach der Operation allmählich nachlassen und zu einem erneuten Ptosis führen. Besonders bei Kindern ist aufgrund des Wachstums häufig eine erneute Operation erforderlich. Auch das bei der Stirnaufhängung verwendete Material kann langfristig an Zugkraft verlieren. Regelmäßige Nachuntersuchungen sind wichtig.
Das Wesen der CPEO ist eine Störung der oxidativen Phosphorylierung aufgrund von mtDNA- oder nDNA-Mutationen.
Mitochondrien besitzen ihre eigene DNA (mtDNA), die 13 für die oxidative Phosphorylierung notwendige Proteine kodiert. Deletionen oder Punktmutationen der mtDNA verringern die Aktivität der Enzyme der Atmungskette und führen zu einem ATP-Mangel. Die äußeren Augenmuskeln haben im Vergleich zur Skelettmuskulatur einen höheren Mitochondriengehalt und einen größeren Stoffwechselbedarf, um die Ermüdungsresistenz aufrechtzuerhalten. Diese Eigenschaft wird als einer der Gründe dafür angesehen, dass die äußeren Augenmuskeln bei CPEO selektiv betroffen sind.
Heteroplasmie ist ein wichtiges pathophysiologisches Konzept der CPEO. Es beschreibt das Vorhandensein von normaler und mutierter mtDNA in derselben Zelle. Wenn der Anteil der mutierten mtDNA einen Schwellenwert (normalerweise >80%) überschreitet, wird das Mitochondrium dysfunktional.
Visuttijai et al. (2021) berichteten über zwei neue Punktmutationen im MT-TN-Gen (m.5669G>A und m.5702delA). Die Einzelfaseranalyse zeigte, dass der Mutationsanteil in COX-negativen Fasern durchschnittlich 93% betrug, signifikant unterschiedlich von COX-normalen Fasern (32% und 57%) (P < 0,0001). Der Schwellenwert für die COX-Dysfunktion lag in beiden Fällen über 80% 2).
Katayama Ueda et al. (2025) identifizierten eine neue Mutation im tRNAGlu-Gen (m.14677T>C) bei einem japanischen Mann mit CPEO. Der mediane Mutationsanteil in ragged-red-Fasern betrug 88,1%, signifikant höher als in nicht-ragged-red-Fasern (Median 17,1%) (P = 0,03) 3).
Hinsichtlich nDNA-Mutationen ist das POLG-Gen eine der häufigsten nukleären genetischen Ursachen der CPEO. POLG kodiert die für die mtDNA-Replikation notwendige DNA-Polymerase γ, und seine Mutationen führen über eine Störung der mtDNA-Replikation zu multiplen mtDNA-Deletionen.
Liu et al. (2023) berichteten über eine 38-jährige Frau mit einer bekannten Mutation c.2857C>T (p.R953C) und einer neuen Mutation c.2391G>C (p.M797I) im POLG-Gen. Sie entwickelte Schwäche und Taubheit der Gliedmaßen sowie Ptosis, und eine Muskelbiopsie zeigte ragged-red-Fasern 5).
7. Aktuelle Forschung und Zukunftsperspektiven (Forschungsstadium-Berichte)
Bei der im Kindesalter beginnenden mitochondrialen Enzephalomyopathie wurde vorgeschlagen, dass die Kombinationstherapie mit 5-Aminolävulinsäure (5-ALA) und Eisen die ATP-Produktion steigert, und klinische Studien wurden begonnen. Die Wirksamkeit bei CPEO ist jedoch unbekannt.
KH176 ist ein mitochondrial gezielter Redoxregulator, der die durch reaktive Sauerstoffspezies vermittelte Zellschädigung reduziert.
Eine klinische Phase-II-Studie zur Bewertung der Sicherheit und Wirksamkeit von KH176 (100 mg zweimal täglich) bei Patienten mit CPEO wurde gestartet (NCT04604548)5).
Es wird versucht, ein normales Gen mit einem sicheren Vektor einzuführen. Darüber hinaus wurde eine Methode berichtet, bei der mutierte mtDNA unter Ausnutzung der Heteroplasmie-Eigenschaften mit Oligo-RNA unterdrückt wird.
Als mitochondriale Genom-Manipulationstechniken wird die selektive Entfernung mutierter mtDNA durch Restriktionsenzyme, TALEN, ZFN und CRISPR untersucht1). Die Keimbahn-Ersatztherapie (Vorkerntransfer, Eizellspindeltransfer) befindet sich ebenfalls im Stadium von Tiermodellen und klinischen Studien.
Fan et al. (2021) detektierten in der Muskel-MR-Spektroskopie von Patienten mit CPEO-plus-Syndrom einen Doppelpeak bei 1-2 ppm (was auf Laktatakkumulation hindeutet). Dieser war in ödematösen Muskeln ausgeprägter, was auf sein Potenzial als Biomarker für die Krankheitsüberwachung hinweist4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.