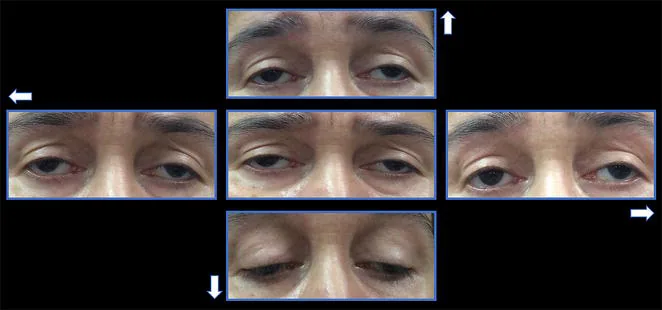
Liệt vận nhãn ngoài tiến triển mạn tính (chronic progressive external ophthalmoplegia: CPEO) là một dạng bệnh toàn thân (bệnh não cơ ty thể) do rối loạn chức năng ty thể. Bệnh ảnh hưởng chọn lọc đến các cơ ngoài nhãn cầu, gây sụp mi hai bên tiến triển chậm và hạn chế vận nhãn.
Đây được coi là kiểu hình phổ biến nhất của bệnh ty thể. Theo cơ sở dữ liệu đoàn hệ tại Anh, tỷ lệ hiện mắc khoảng 1/30.000 và tỷ lệ mới mắc 1-2/100.000 1).
Lịch sử CPEO bắt đầu với mô tả đầu tiên của von Graefe năm 1868. Năm 1958, Kearns và Sayre mô tả bộ ba CPEO, viêm võng mạc sắc tố và rối loạn dẫn truyền tim. Năm 1972, sợi cơ đỏ rách (ragged red fiber) được phát hiện trên sinh thiết cơ, và năm 1988-1989 phát hiện mất đoạn mtDNA. Năm 2000, đột biến DNA nhân (nDNA) đầu tiên liên quan đến mất đoạn mtDNA đa ổ được xác định 1).
Về mặt lâm sàng, CPEO được chia thành hai loại sau.
CPEO đơn độc: Chỉ có triệu chứng ở mắt, không có dấu hiệu toàn thân. Khởi phát điển hình ở độ tuổi 30-40.
CPEO-plus: Kèm theo các triệu chứng toàn thân như mất thính lực thần kinh giác quan, khó nuốt, rối loạn dẫn truyền tim, mất điều hòa tiểu não, yếu cơ gốc chi. Hội chứng Kearns-Sayre (KSS) là bệnh điển hình.
Một số trường hợp khởi phát trước hoặc sau tuổi dậy thì, ít có dấu hiệu lâm sàng toàn thân, thường đến khám bác sĩ nhãn khoa trước tiên.
QSự khác biệt giữa CPEO và CPEO-plus là gì?
A
CPEO đơn độc chỉ bao gồm sụp mi và rối loạn vận động mắt mà không có triệu chứng toàn thân. CPEO-plus kèm theo các triệu chứng toàn thân như mất thính lực thần kinh giác quan, khó nuốt, rối loạn dẫn truyền tim, mất điều hòa tiểu não. Hội chứng Kearns-Sayre là bệnh CPEO-plus điển hình khởi phát trước 20 tuổi, kèm viêm võng mạc sắc tố và rối loạn dẫn truyền tim.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
QTại sao bệnh nhân CPEO ít khi nhận thấy song thị?
A
Vì CPEO ảnh hưởng cả hai mắt đối xứng và từ từ, sự khác biệt vận động mắt nhỏ nên song thị hiếm khi xảy ra. Ngoài ra, tiến triển rất chậm cho phép thích nghi trung ương. Chỉ khoảng một phần ba bệnh nhân có song thị liên tục hoặc từng lúc.
Nguyên nhân CPEO chủ yếu được chia thành đột biến DNA ty thể (mtDNA) và đột biến DNA nhân (nDNA).
Đột biến mtDNA
Mất đoạn lớn đơn độc: Nguyên nhân phổ biến nhất, chiếm 60-80% trường hợp. Phần lớn là mất đoạn trong khoảng 1,3-1,9 kb. Các trường hợp lẻ tẻ gợi ý đột biến de novo.
Đột biến điểm: Gen tRNA là điểm nóng. Ít nhất 35 đột biến điểm có thể biểu hiện dưới dạng CPEO đơn độc. Gen MT-TN (tRNAAsn) là điểm nóng cho PEO lẻ tẻ, với 7 trong số 11 đột biến cho kiểu hình PEO 2).
Kiểu di truyền: Di truyền theo dòng mẹ. Dị hợp thể (hỗn hợp mtDNA bình thường và mtDNA đột biến) là đặc trưng, và khi tỷ lệ đột biến vượt quá 80%, rối loạn chức năng ở cấp độ tế bào trở nên rõ rệt2)3).
Đột biến nDNA
Các gen liên quan đến duy trì mtDNA: Đột biến đã được báo cáo ở POLG (DNA polymerase γ), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK và các gen khác1)5).
Mất đoạn mtDNA nhiều nơi: Đột biến nDNA gây ra mất đoạn mtDNA nhiều nơi thông qua sự suy giảm sao chép và sửa chữa mtDNA.
Kiểu di truyền: Di truyền trội trên nhiễm sắc thể thường hoặc lặn trên nhiễm sắc thể thường. Việc hỏi tiền sử gia đình rất quan trọng.
Lý do các cơ ngoại nhãn bị ảnh hưởng chọn lọc là vì chúng có hàm lượng ty thể cao hơn và nhu cầu trao đổi chất lớn hơn so với cơ xương, khiến chúng dễ bị tổn thương hơn đối với rối loạn phosphoryl hóa oxy hóa.
QCPEO có di truyền không?
A
Kiểu di truyền khác nhau tùy theo gen gây bệnh. Các trường hợp lẻ tẻ đột biến mtDNA là đột biến mới và nguy cơ di truyền thấp. Các trường hợp đột biến nDNA biểu hiện di truyền trội hoặc lặn trên nhiễm sắc thể thường, với sự xuất hiện trong gia đình. Đột biến mtDNA cũng có thể lây truyền qua di truyền dòng mẹ. Tư vấn di truyền được khuyến nghị.
CPEO được chẩn đoán chủ yếu dựa trên lâm sàng, nhưng các xét nghiệm khác nhau rất hữu ích để xác định chẩn đoán và chẩn đoán phân biệt. Tại các phòng khám chuyên khoa, sự chú ý có thể tập trung vào một bệnh cụ thể, dẫn đến bỏ sót sụp mi hoặc rối loạn vận động mắt 6).
Xét nghiệm máu: Có thể thấy tăng lactate huyết thanh, creatine kinase (CK) và lactate dịch não tủy, nhưng độ nhạy và độ đặc hiệu không cao. Tăng pyruvate máu hoặc tỷ lệ lactate/pyruvate cũng có giá trị tham khảo.
Xét nghiệm kháng thể: Xác nhận âm tính với kháng thể thụ thể acetylcholine và tự kháng thể tuyến giáp hữu ích để loại trừ nhược cơ và bệnh mắt do tuyến giáp.
Dấu ấn sinh học: Yếu tố tăng trưởng nguyên bào sợi 21 (FGF-21) và yếu tố biệt hóa tăng trưởng 15 (GDF-15) đang được chú ý như một phương pháp sàng lọc không xâm lấn 1).
Trên điện cơ của cơ ngoại nhãn, thấy dạng giao thoa với phóng điện đủ mặc dù biên độ yếu hơn cơ bình thường, ngay cả khi không thể cử động mắt. Điều này hữu ích để phân biệt với bệnh thần kinh.
Mất đoạn mtDNA: Phương pháp Southern blot sử dụng mẫu sinh thiết cơ xương là đáng tin cậy. PCR dài có thể phát hiện trong máu ngoại vi trong một số trường hợp.
Đột biến điểm mtDNA: Phân tích toàn bộ mtDNA bằng giải trình tự thế hệ mới (NGS) rất hiệu quả. Đôi khi không phát hiện được trong máu mà chỉ phát hiện trong mô cơ 2)3).
Đột biến nDNA: Phân tích exome các gen liên quan như POLG, TWNK, SLC25A4 được thực hiện 5).
MRI não: Có thể thấy tín hiệu cao chất trắng, teo vỏ não, teo tiểu não, và tín hiệu cao thân não. Trong một trường hợp KSS, đã có báo cáo về teo tiểu não một bên tiến triển theo thời gian 7).
Phổ MR (MRS): Có thể phát hiện không xâm lấn sự tích tụ lactate trong cơ, và có thể hữu ích cho theo dõi bệnh 4).
Bệnh nhược cơ là bệnh chẩn đoán phân biệt quan trọng nhất vì gây sụp mí và rối loạn vận động mắt. Bệnh nhược cơ đặc trưng bởi dao động trong ngày và dễ mệt mỏi, cải thiện với nghiệm pháp Tensilon và nghiệm pháp chườm đá. Sụp mí trong CPEO không dao động và không cải thiện với các nghiệm pháp này.
Kính lăng kính: Hiệu quả trong việc giảm song thị cho các lệch mắt góc nhỏ.
Phẫu thuật lác: Rút ngắn cơ gây rối loạn vận động nhãn cầu để chỉnh ở vị trí nguyên phát. Nếu góc lớn, cũng thực hiện lùi cơ đối vận. Do bệnh tiến triển, có khả năng tái phát.
Đánh giá chức năng tim: Trong KSS, có nguy cơ rối loạn dẫn truyền tim (block nhĩ thất), cần điện tâm đồ định kỳ và quyết định đặt máy tạo nhịp.
Rối loạn nội tiết: Cần quản lý đái tháo đường, thiếu hụt hormone tăng trưởng và thấp lùn.
Đánh giá thính lực: Khuyến cáo kiểm tra thính lực định kỳ cho điếc thần kinh giác quan.
QSụp mi có thể tái phát sau phẫu thuật không?
A
Vì CPEO là bệnh tiến triển, chức năng cơ nâng mi có thể xấu dần sau phẫu thuật, dẫn đến tái phát sụp mi. Đặc biệt ở trẻ em, thường cần phẫu thuật lại khi lớn lên. Vật liệu dùng trong phẫu thuật treo mi lên cơ trán có thể giảm lực kéo về lâu dài. Theo dõi định kỳ rất quan trọng.
Bản chất của CPEO là rối loạn phosphoryl hóa oxy hóa do đột biến mtDNA hoặc nDNA.
Ty thể có DNA riêng (mtDNA) mã hóa 13 protein cần thiết cho quá trình phosphoryl hóa oxy hóa. Mất đoạn hoặc đột biến điểm trong mtDNA làm giảm hoạt động của các enzyme chuỗi vận chuyển điện tử, gây thiếu hụt sản xuất ATP. Các cơ ngoại nhãn có hàm lượng ty thể cao hơn so với cơ xương và có nhu cầu chuyển hóa lớn hơn để duy trì khả năng chống mệt mỏi. Đặc tính này được coi là một trong những lý do khiến các cơ ngoại nhãn bị tổn thương chọn lọc trong CPEO.
Dị hợp tử (Heteroplasmy) là một khái niệm bệnh lý quan trọng trong CPEO. Đó là hiện tượng mtDNA bình thường và mtDNA đột biến cùng tồn tại trong một tế bào. Khi tỷ lệ mtDNA đột biến vượt quá ngưỡng (thường >80%), ty thể đó trở nên rối loạn chức năng.
Visuttijai và cộng sự (2021) đã báo cáo hai đột biến điểm mới trong gen MT-TN (m.5669G>A và m.5702delA). Phân tích sợi cơ đơn cho thấy tỷ lệ đột biến trung bình trong các sợi COX-âm tính là 93%, khác biệt có ý nghĩa so với các sợi COX-bình thường (32% và 57%) (P < 0,0001). Ngưỡng rối loạn chức năng COX trong cả hai trường hợp là >80%2).
Katayama Ueda và cộng sự (2025) đã xác định một đột biến mới (m.14677T>C) trong gen tRNAGlu ở một trường hợp CPEO nam người Nhật. Tỷ lệ đột biến trung vị trong các sợi đỏ ráp (ragged red fibers) là 88,1%, cao hơn đáng kể so với các sợi không ráp (trung vị 17,1%) (P = 0,03)3).
Về đột biến nDNA, gen POLG là một trong những nguyên nhân nhân phổ biến nhất của CPEO. POLG mã hóa DNA polymerase γ cần thiết cho sự sao chép mtDNA, và các đột biến của nó gây ra mất đoạn nhiều mtDNA thông qua suy giảm sao chép mtDNA.
Liu và cộng sự (2023) đã báo cáo một phụ nữ 38 tuổi có đột biến đã biết c.2857C>T (p.R953C) và đột biến mới c.2391G>C (p.M797I) trong gen POLG. Sụp mí mắt xuất hiện ngoài yếu và tê ở tứ chi, và sinh thiết cơ xác nhận có sợi đỏ ráp5).
7. Nghiên cứu mới nhất và Triển vọng tương lai (Báo cáo giai đoạn nghiên cứu)
Trong bệnh não cơ ty thể khởi phát ở trẻ em, liệu pháp kết hợp axit 5-aminolevulinic (5-ALA) và bổ sung sắt đã được gợi ý để tăng sản xuất ATP, và các thử nghiệm lâm sàng đã được bắt đầu. Tuy nhiên, hiệu quả của nó trong CPEO vẫn chưa được biết.
KH176 là một chất điều chỉnh oxy hóa khử nhắm vào ty thể, làm giảm tổn thương tế bào qua trung gian các loại oxy phản ứng.
Một thử nghiệm lâm sàng Giai đoạn II đánh giá tính an toàn và hiệu quả của KH176 (100 mg hai lần mỗi ngày) ở bệnh nhân CPEO đã được bắt đầu (NCT04604548)5).
Các nỗ lực đang được thực hiện để đưa gen bình thường vào bằng vector an toàn. Một phương pháp ức chế mtDNA đột biến bằng oligo RNA, tận dụng đặc tính dị hợp tử, cũng đã được báo cáo.
Là các kỹ thuật thao tác bộ gen ty thể, việc loại bỏ chọn lọc mtDNA đột biến bằng enzyme giới hạn, TALEN, ZFN và CRISPR đang được nghiên cứu1). Liệu pháp thay thế dòng mầm (chuyển nhân tiền nhân/chuyển thoi noãn) cũng đang ở giai đoạn mô hình động vật và thử nghiệm lâm sàng.
Fan và cộng sự (2021) đã phát hiện đỉnh kép ở 1-2 ppm (gợi ý tích tụ lactate) trên phổ MR cơ ở bệnh nhân hội chứng CPEO-plus. Nó nổi bật hơn ở cơ phù nề, cho thấy tiềm năng như một dấu ấn sinh học để theo dõi bệnh4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.