L’oftalmoplegia esterna progressiva cronica (CPEO) è una forma di encefalomiopatia mitocondriale, un gruppo di malattie sistemiche dovute a disfunzione mitocondriale. Colpisce selettivamente i muscoli extraoculari, causando ptosi bilaterale lentamente progressiva e limitazione dei movimenti oculari.
È il fenotipo più frequente delle malattie mitocondriali. Secondo un database di coorte britannico, la prevalenza è di circa 1/30.000 e l’incidenza di 1-2/100.0001).
La storia della CPEO inizia con la prima descrizione di von Graefe nel 1868. Nel 1958 Kearns e Sayre descrissero la triade CPEO, retinite pigmentosa e disturbo della conduzione cardiaca. Nel 1972 furono scoperte le fibre rosse sfilacciate (ragged red fibers) alla biopsia muscolare, e nel 1988-1989 furono rilevate delezioni del mtDNA. Nel 2000 fu identificata la prima mutazione del DNA nucleare (nDNA) associata a delezioni multiple del mtDNA1).
Clinicamente, la CPEO è suddivisa in due tipi principali.
CPEO isolato: solo sintomi oculari, senza segni sistemici. L’esordio tipico è tra i 30 e i 40 anni.
CPEO-plus: accompagnato da sintomi sistemici come ipoacusia neurosensoriale, disfagia, disturbi della conduzione cardiaca, atassia cerebellare, debolezza muscolare prossimale. La sindrome di Kearns-Sayre (KSS) ne è un esempio rappresentativo.
Ci sono anche casi con esordio intorno alla pubertà, con pochi segni clinici sistemici, e i pazienti spesso consultano prima un oculista.
QQual è la differenza tra CPEO e CPEO-plus?
A
Il CPEO isolato presenta solo ptosi e disturbi oculomotori senza sintomi sistemici. Il CPEO-plus associa sintomi sistemici come ipoacusia neurosensoriale, disfagia, disturbi della conduzione cardiaca, atassia cerebellare. La sindrome di Kearns-Sayre è una malattia rappresentativa del CPEO-plus, con esordio prima dei 20 anni, retinite pigmentosa e disturbi della conduzione cardiaca.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
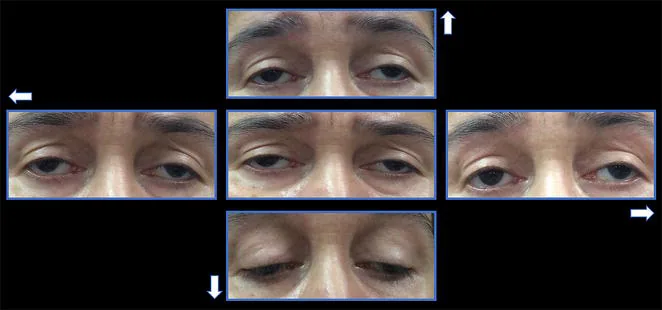
La comparsa dei sintomi del CPEO è estremamente lenta. Il primo sintomo è spesso la ptosi.
Ptosi: il paziente si presenta con la lamentela che «la palpebra non si solleva bene». Progredisce gradualmente da unilaterale a bilaterale. Non è raro che il paziente non se ne accorga finché qualcun altro non glielo fa notare.
Disturbi oculomotori: compaiono alcuni anni dopo la ptosi. Una limitazione del movimento in tutte le direzioni progredisce gradualmente.
Assenza di diplopia: poiché entrambi gli occhi sono colpiti simmetricamente, i pazienti spesso non avvertono diplopia nonostante marcati disturbi oculomotori.
Posizione compensatoria della testa: il paziente compensa la mancanza di movimenti oculari con cambiamenti della posizione della testa.
Intolleranza all’esercizio: nei casi con debolezza muscolare generalizzata, il paziente lamenta facile affaticabilità.
L’assenza di dolore è caratteristica della CPEO. Dolore oculare, esoftalmo o anomalie pupillari suggeriscono altre eziologie.
Reperti clinici (osservati dal medico durante l’esame)
Ptosi : bilaterale, simmetrica, con funzione del muscolo elevatore ridotta a meno di 8-10 mm 1). Nei casi avanzati, ptosi grave.
Limitazione dei movimenti oculari : in tutte le direzioni. In posizione primaria può essere presente exotropia.
Conservazione pupillare : la CPEO non si associa ad anomalie pupillari. La dilatazione pupillare suggerisce altre cause, come la paralisi del nervo oculomotore.
Incompleta chiusura palpebrale : associata alla ptosi a causa della debolezza del muscolo orbicolare, che può portare a cheratite da esposizione.
Reperti retinici : all’angiografia con fluoresceina può essere presente iperfluorescenza granulare a livello dell’epitelio pigmentato retinico. Una retinopatia a sale e pepe suggerisce la sindrome di Kearns-Sayre.
QPerché nella CPEO la diplopia è raramente avvertita?
A
Nella CPEO entrambi gli occhi sono colpiti simmetricamente e lentamente, quindi la differenza nei movimenti oculari è ridotta e la diplopia è rara. Inoltre, la progressione molto lenta consente un adattamento centrale. Solo circa un terzo dei pazienti presenta diplopia costante o intermittente.
Le cause della CPEO si dividono in mutazioni del DNA mitocondriale (mtDNA) e mutazioni del DNA nucleare (nDNA).
Mutazioni del mtDNA
Delezione singola di grandi dimensioni : causa più comune, rappresenta il 60-80% dei casi. La maggior parte delle delezioni è compresa tra 1,3 e 1,9 kb. I casi sporadici suggeriscono una mutazione de novo.
Mutazioni puntiformi : i geni tRNA sono hot spot. Almeno 35 mutazioni puntiformi possono manifestarsi come CPEO isolata. Il gene MT-TN (tRNAAsn) è un hot spot per la PEO sporadica e 7 delle 11 mutazioni presentano un fenotipo PEO 2).
Modalità di trasmissione : eredità materna. L’eteroplasmia (miscela di mtDNA normale e mutato) è caratteristica; quando la percentuale di mutazione supera l’80%, si manifesta una disfunzione a livello cellulare2)3).
Mutazione del DNA nucleare
Geni coinvolti nel mantenimento del mtDNA : sono state riportate mutazioni in POLG (DNA polimerasi γ), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK e altri1)5).
Delezioni multiple del mtDNA : le mutazioni del DNA nucleare causano delezioni multiple del mtDNA attraverso l’alterazione della replicazione e riparazione del mtDNA.
Modalità di trasmissione : autosomica dominante o autosomica recessiva. È importante raccogliere la storia familiare.
Il motivo per cui i muscoli extraoculari sono selettivamente colpiti è che, rispetto ai muscoli scheletrici, hanno un contenuto mitocondriale più elevato e un maggiore fabbisogno metabolico, rendendoli più vulnerabili ai disturbi della fosforilazione ossidativa.
QLa CPEO è ereditaria?
A
La modalità di trasmissione varia a seconda del gene causale. I casi sporadici di mutazione del mtDNA sono mutazioni de novo e il rischio genetico è basso. I casi con mutazione del DNA nucleare mostrano ereditarietà autosomica dominante o recessiva, con casi familiari. Anche le mutazioni del mtDNA possono essere trasmesse per via materna. Si raccomanda la consulenza genetica.
La CPEO si basa principalmente sulla diagnosi clinica, ma vari esami sono utili per la conferma e la diagnosi differenziale. Nelle cliniche specialistiche l’attenzione può concentrarsi su una specifica malattia e la ptosi o i disturbi dei movimenti oculari possono essere trascurati 6).
Esami del sangue : Possono essere osservati un aumento del lattato sierico, della creatin chinasi (CK) e del lattato nel liquido cerebrospinale, ma la sensibilità e la specificità non sono elevate. Anche un aumento del piruvato nel sangue e del rapporto lattato/piruvato può essere utile.
Test anticorpali : La conferma della negatività degli anticorpi anti-recettore dell’acetilcolina e degli autoanticorpi tiroidei è utile per escludere la miastenia grave e l’oftalmopatia tiroidea.
Biomarcatori : Il fattore di crescita dei fibroblasti 21 (FGF-21) e il fattore di differenziazione della crescita 15 (GDF-15) stanno attirando l’attenzione come screening non invasivo 1).
Questo è l’esame chiave per la diagnosi definitiva.
Colorazione di Gomori al tricromio : Si osservano fibre rosse sfilacciate (ragged red fibers). I mitocondri accumulati all’interno o intorno alle fibre muscolari vengono colorati intensamente.
Doppia colorazione COX/SDH : Le fibre negative per la citocromo c ossidasi (COX) appaiono a mosaico.
Microscopia elettronica : Si osservano mitocondri giganti e morfologicamente anomali tra le fibre muscolari.
L’elettromiografia dei muscoli extraoculari mostra un pattern di interferenza con ampiezza leggermente inferiore rispetto ai muscoli normali, ma con scarica sufficiente anche in assenza di movimenti oculari. Utile per la diagnosi differenziale con le malattie neurogene.
Delezione del mtDNA : Il metodo Southern blot su campione di biopsia muscolare scheletrica è affidabile. Con il metodo long PCR può talvolta essere rilevato anche nel sangue periferico.
Mutazione puntiforme del mtDNA : L’analisi completa del mtDNA mediante sequenziamento di nuova generazione (NGS) è efficace. Talvolta è rilevabile solo nel tessuto muscolare e non nel sangue 2)3).
Mutazione del DNA nucleare: viene eseguita l’analisi dell’esoma dei geni correlati come POLG, TWNK, SLC25A4 5).
RMN cerebrale: può mostrare iperintensità della sostanza bianca, atrofia corticale, atrofia cerebellare, iperintensità del tronco encefalico, ecc. Un caso di KSS ha riportato atrofia cerebellare unilaterale progressiva nel tempo 7).
Spettroscopia RM (MRS): può rilevare in modo non invasivo l’accumulo di lattato nei muscoli, potenzialmente utile per il monitoraggio della malattia 4).
Atrofia muscolare a prevalenza distale. Miotonia da percussione
La miastenia gravis è la più importante diagnosi differenziale perché causa ptosi e disturbi dei movimenti oculari. Nella miastenia gravis sono caratteristiche le fluttuazioni diurne e l’affaticabilità, e si osserva un miglioramento con il test del Tensilon o il test del ghiaccio. La ptosi della CPEO non è fluttuante e non migliora con questi test.
Non esiste una terapia curativa stabilita per la CPEO. Il mantenimento della qualità della vita (QdV) con terapia sintomatica è il cardine del trattamento.
Indicazione : quando la funzione del muscolo elevatore è conservata in modo moderato o buono.
Metodo : si esegue l’avanzamento o la resezione del muscolo elevatore della palpebra superiore. Per via percutanea si avanza e si fissa l’aponeurosi, con eventuale tucking del muscolo di Müller.
Nota : nella CPEO la funzione dell’elevatore peggiora progressivamente, quindi l’effetto può diminuire a lungo termine.
Sospensione al muscolo frontale
Indicazioni: quando la funzione del muscolo elevatore è scarsa (meno di 4 mm). Nella CPEO spesso si sceglie questa opzione.
Metodo: connessione della palpebra superiore al muscolo frontale utilizzando fascia autologa (fascia lata, fascia temporale), foglio di Gore-Tex®, filo di nylon, asta di silicone, ecc.
Una valutazione preoperatoria accurata da parte di un chirurgo oculoplastico esperto è indispensabile. In assenza del fenomeno di Bell, è opportuno mirare a una ipocorrezione.
Lenti prismatiche: efficaci per ridurre la diplopia in caso di piccoli angoli di deviazione oculare.
Chirurgia dello strabismo: accorciamento del muscolo oculomotore deficitario per correggere la posizione primaria. Se l’angolo è grande, si associa anche la recessione del muscolo antagonista. A causa della natura progressiva della malattia, è possibile una recidiva.
Valutazione cardiaca: nella KSS esiste il rischio di disturbi della conduzione cardiaca (blocco atrioventricolare); sono necessari ECG regolari e la decisione di impiantare un pacemaker.
Anomalie endocrine: è richiesta la gestione del diabete, del deficit dell’ormone della crescita, della bassa statura, ecc.
Valutazione uditiva: si raccomandano esami audiometrici regolari per l’ipoacusia neurosensoriale.
QIl ptosi può recidivare dopo l'intervento chirurgico?
A
La CPEO è una malattia progressiva, quindi anche dopo l’intervento la funzione dell’elevatore può peggiorare gradualmente, portando a una recidiva del ptosi. Soprattutto nei bambini, con la crescita è spesso necessario un reintervento. Anche i materiali utilizzati per la sospensione frontale possono perdere forza di trazione a lungo termine. È importante un follow-up regolare.
6. Fisiopatologia e meccanismi dettagliati di insorgenza
L’essenza della CPEO è un disturbo della fosforilazione ossidativa dovuto a mutazioni del mtDNA o del nDNA.
I mitocondri possiedono un proprio DNA (mtDNA) che codifica 13 proteine necessarie per la fosforilazione ossidativa. Delezioni o mutazioni puntiformi del mtDNA riducono l’attività degli enzimi della catena di trasporto degli elettroni, portando a una produzione insufficiente di ATP. I muscoli extraoculari hanno un contenuto mitocondriale più elevato rispetto ai muscoli scheletrici e un fabbisogno metabolico maggiore per mantenere la resistenza all’affaticamento. Questa caratteristica è considerata una delle ragioni per cui i muscoli extraoculari sono selettivamente colpiti nella CPEO.
L’eteroplasmia è un importante concetto fisiopatologico della CPEO. È il fenomeno della coesistenza di mtDNA normale e mutato all’interno di una singola cellula. Quando la proporzione di mtDNA mutato supera una soglia (di solito >80%), il mitocondrio diventa disfunzionale.
Visuttijai et al. (2021) hanno riportato due nuove mutazioni puntiformi nel gene MT-TN (m.5669G>A e m.5702delA). L’analisi di singole fibre muscolari ha mostrato che la percentuale media di mutazione nelle fibre COX-negative era rispettivamente del 93%, significativamente diversa dalle fibre COX-normali (32% e 57%) (P < 0,0001). La soglia per la disfunzione della COX era superiore all’80% in entrambi i casi 2).
Katayama Ueda et al. (2025) hanno identificato una nuova mutazione nel gene tRNAGlu (m.14677T>C) in un uomo giapponese con CPEO. La percentuale mediana di mutazione nelle fibre ragged-red era dell’88,1%, significativamente più alta rispetto alle fibre non ragged-red (mediana 17,1%) (P = 0,03) 3).
Per quanto riguarda le mutazioni del nDNA, il gene POLG è una delle cause genetiche nucleari più comuni della CPEO. POLG codifica per la DNA polimerasi γ necessaria per la replicazione del mtDNA, e le sue mutazioni portano a delezioni multiple del mtDNA attraverso un disturbo della replicazione.
Liu et al. (2023) hanno riportato il caso di una donna di 38 anni con una mutazione nota c.2857C>T (p.R953C) e una nuova mutazione c.2391G>C (p.M797I) nel gene POLG. Ha sviluppato debolezza e intorpidimento degli arti insieme a ptosi, e una biopsia muscolare ha rivelato fibre ragged-red 5).
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Nell’encefalomiopatia mitocondriale ad esordio infantile, la terapia combinata di acido 5-aminolevulinico (5-ALA) e ferro è stata suggerita per aumentare la produzione di ATP, e sono stati avviati studi clinici. Tuttavia, la sua efficacia nella CPEO è sconosciuta.
KH176 è un agente regolatore redox mirato ai mitocondri, che riduce il danno cellulare mediato dalle specie reattive dell’ossigeno.
È stato avviato uno studio clinico di fase II per valutare la sicurezza e l’efficacia di KH176 (100 mg due volte al giorno) in pazienti con CPEO (NCT04604548)5).
Si stanno tentando approcci per introdurre un gene normale utilizzando un vettore sicuro. Inoltre, è stato riportato un metodo che sfrutta le caratteristiche dell’eteroplasmia per sopprimere il mtDNA mutante con oligo RNA.
Come tecniche di manipolazione del genoma mitocondriale, è in fase di studio la rimozione selettiva del mtDNA mutante mediante enzimi di restrizione, TALEN, ZFN e CRISPR1). La terapia sostitutiva a livello della linea germinale (trasferimento del pronucleo, trasferimento del fuso dell’ovocita) è anch’essa in fase di modelli animali e studi clinici.
Fan et al. (2021) hanno rilevato un doppio picco a 1-2 ppm (suggestivo di accumulo di lattato) alla spettroscopia RM muscolare in pazienti con sindrome CPEO-plus. Tale picco era più pronunciato nei muscoli edematosi, indicando il suo potenziale come biomarcatore per il monitoraggio della malattia4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.