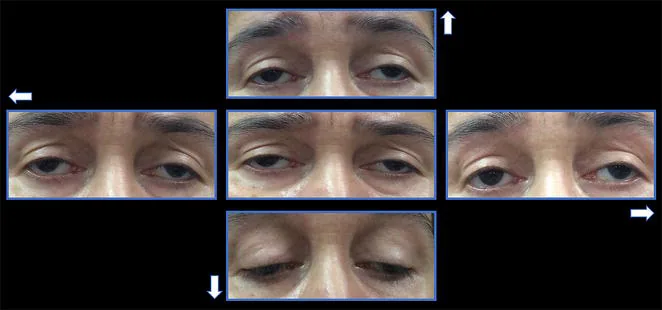
La oftalmoplejía externa progresiva crónica (CPEO) es un tipo de encefalomiopatía mitocondrial, un grupo de enfermedades sistémicas causadas por disfunción mitocondrial. Afecta selectivamente los músculos extraoculares, provocando ptosis bilateral lentamente progresiva y limitación de los movimientos oculares.
Se considera el fenotipo más frecuente entre las enfermedades mitocondriales. Según una base de datos de cohortes del Reino Unido, la prevalencia es de aproximadamente 1/30,000 y la incidencia de 1–2/100,000 1).
La historia de la CPEO comienza con la primera descripción de von Graefe en 1868. En 1958, Kearns y Sayre describieron la tríada de CPEO, retinitis pigmentaria y defectos de conducción cardíaca. En 1972, se encontraron fibras rojas rasgadas en la biopsia muscular, y en 1988–1989 se detectaron deleciones del ADNmt. En 2000, se identificó la primera mutación del ADN nuclear (ADNn) asociada con deleciones múltiples del ADNmt 1).
Clínicamente, la CPEO se divide ampliamente en los siguientes dos tipos.
CPEO aislado: Solo síntomas oculares, sin signos sistémicos. El inicio típico ocurre entre los 30 y 40 años.
CPEO-plus: Acompañado de síntomas sistémicos como hipoacusia neurosensorial, disfagia, trastornos de la conducción cardíaca, ataxia cerebelosa y debilidad muscular proximal. El síndrome de Kearns-Sayre (KSS) es un ejemplo representativo.
Algunos casos se presentan alrededor de la pubertad y, debido a la falta de signos clínicos sistémicos, a menudo acuden primero al oftalmólogo.
Q¿Cuál es la diferencia entre CPEO y CPEO-plus?
A
El CPEO aislado presenta solo ptosis y oftalmoplejía, sin síntomas sistémicos. El CPEO-plus se asocia con síntomas sistémicos como hipoacusia neurosensorial, disfagia, trastornos de la conducción cardíaca y ataxia cerebelosa. El síndrome de Kearns-Sayre es una enfermedad CPEO-plus representativa que comienza antes de los 20 años e incluye retinosis pigmentaria y trastornos de la conducción cardíaca.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
La aparición de los síntomas del CPEO es extremadamente gradual. El síntoma inicial suele ser la ptosis.
Ptosis: Los pacientes consultan por “sensación de que el párpado no se eleva bien”. Progresa gradualmente de unilateral a bilateral. No es raro que el paciente no lo note hasta que alguien se lo señala.
Oftalmoplejía: Aparece varios años después de la ptosis. La limitación del movimiento ocular en todas las direcciones progresa gradualmente.
Ausencia de diplopía: Debido a que ambos ojos están afectados simétricamente, los pacientes a menudo no notan diplopía incluso con oftalmoplejía marcada.
Postura compensatoria de la cabeza: El paciente compensa la limitación del movimiento ocular girando la cabeza.
Intolerancia al ejercicio: Los pacientes con debilidad muscular generalizada se quejan de fatigabilidad fácil.
La ausencia de dolor es una característica de la CPEO. El dolor ocular, la proptosis o las anomalías pupilares sugieren otras etiologías.
Hallazgos clínicos (hallazgos confirmados por el médico en la exploración)
Ptosis: Bilateral, simétrica, con función del elevador reducida a menos de 8-10 mm 1). En casos avanzados, se produce ptosis grave.
Restricción del movimiento ocular: En todas las direcciones. Puede haber exotropía en la posición primaria.
Preservación pupilar: La CPEO no se asocia con anomalías pupilares. Si hay midriasis, considere otras causas como parálisis del nervio oculomotor.
Lagoftalmos: Junto con la ptosis, la debilidad del músculo orbicular de los párpados provoca un cierre incompleto del párpado, que puede causar queratitis por exposición.
Hallazgos retinianos: La angiografía con fluoresceína puede mostrar hiperfluorescencia granular a nivel del epitelio pigmentario de la retina. Si se acompaña de retinopatía en sal y pimienta, sugiere el síndrome de Kearns-Sayre.
Q¿Por qué la diplopía es menos perceptible en la CPEO?
A
Debido a que la CPEO afecta ambos ojos de forma simétrica y lenta, hay poca diferencia en el movimiento ocular entre los dos ojos, lo que hace que la diplopía sea menos probable. Además, debido a la progresión muy lenta, se produce una adaptación central. Solo aproximadamente un tercio de los pacientes experimentan diplopía constante o intermitente.
Las causas de la CPEO se dividen ampliamente en mutaciones del ADN mitocondrial (ADNmt) y mutaciones del ADN nuclear (ADNn).
Mutaciones del ADNmt
Deleción única grande: La causa más común, que representa el 60-80% de los casos. La mayoría de las deleciones oscilan entre 1,3 y 1,9 kb. Los casos esporádicos sugieren mutaciones de novo.
Mutaciones puntuales: Los genes de ARNt son puntos calientes. Al menos 35 mutaciones puntuales pueden manifestarse como CPEO aislada. El gen MT-TN (ARNtAsn) es un punto caliente para la PEO esporádica, con 7 de cada 11 mutaciones que presentan un fenotipo PEO 2).
Patrón de herencia: Herencia materna. La heteroplasmia (coexistencia de mtDNA normal y mutante) es característica; cuando la proporción de mtDNA mutante supera el 80%, la disfunción se hace evidente a nivel celular2)3).
Mutaciones en el nDNA
Genes relacionados con el mantenimiento del mtDNA: Se han reportado mutaciones en POLG (ADN polimerasa gamma), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK, entre otros1)5).
Deleciones múltiples del mtDNA: Las mutaciones en el nDNA causan deleciones múltiples del mtDNA a través de la alteración de la replicación y reparación del mtDNA.
Patrón de herencia: Herencia autosómica dominante o autosómica recesiva. Es importante obtener la historia familiar.
La razón por la que los músculos extraoculares se ven afectados selectivamente es que tienen un mayor contenido mitocondrial y una mayor demanda metabólica en comparación con el músculo esquelético, lo que los hace más vulnerables a la alteración de la fosforilación oxidativa.
Q¿La CPEO es hereditaria?
A
El patrón de herencia varía según el gen causante. Los casos esporádicos de mutaciones en el mtDNA son mutaciones de novo con bajo riesgo genético. Los casos con mutaciones en el nDNA muestran herencia autosómica dominante o recesiva y pueden presentarse en familias. Incluso las mutaciones en el mtDNA pueden transmitirse por herencia materna. Se recomienda asesoramiento genético.
La CPEO es principalmente un diagnóstico clínico, pero varias pruebas son útiles para la confirmación y el diagnóstico diferencial. En clínicas especializadas, la atención puede centrarse en enfermedades específicas y la ptosis o los trastornos del movimiento ocular pueden pasarse por alto 6).
Análisis de sangre: El lactato sérico, la creatina quinasa (CK) y el lactato del líquido cefalorraquídeo pueden estar elevados, pero la sensibilidad y especificidad no son altas. La elevación del piruvato en sangre y el aumento de la relación lactato/piruvato también son informativos.
Pruebas de anticuerpos: La negatividad de los anticuerpos contra el receptor de acetilcolina y los autoanticuerpos tiroideos es útil para excluir miastenia gravis y oftalmopatía tiroidea.
Biomarcadores: El factor de crecimiento de fibroblastos 21 (FGF-21) y el factor de diferenciación de crecimiento 15 (GDF-15) están ganando atención como herramientas de detección no invasivas 1).
Esta es la prueba clave para el diagnóstico definitivo.
Tinción de Gomori tricrómica: Muestra fibras rojas rasgadas. Las mitocondrias acumuladas dentro o alrededor de las fibras musculares se tiñen intensamente.
Doble tinción COX/SDH: Muestra una apariencia en mosaico de fibras negativas para citocromo c oxidasa (COX).
Microscopía electrónica: Muestra mitocondrias gigantes con morfología anormal entre las fibras musculares.
La electromiografía de los músculos extraoculares muestra patrones de interferencia con descarga suficiente, aunque con amplitud ligeramente más débil que los músculos normales, incluso cuando no es posible el movimiento ocular. Esto es útil para diferenciar de enfermedades neurogénicas.
Deleción de mtDNA: El análisis de Southern blot con muestras de biopsia de músculo esquelético es fiable. La PCR larga puede permitir la detección en sangre periférica en algunos casos.
Mutaciones puntuales de mtDNA: El análisis completo de mtDNA mediante secuenciación de próxima generación (NGS) es efectivo. Pueden ser indetectables en sangre y solo detectarse en tejido muscular 2)3).
Mutaciones del ADNn: Se realiza un análisis del exoma de genes relacionados como POLG, TWNK y SLC25A4 5).
RM cerebral: Puede mostrar hiperintensidades de la sustancia blanca, atrofia cortical, atrofia cerebelosa e hiperintensidades del tronco encefálico. En un caso de KSS, se informó atrofia cerebelosa unilateral que progresó con el tiempo 7).
Espectroscopia por RM (MRS): Puede detectar de forma no invasiva la acumulación de lactato en los músculos y puede ser útil para el monitoreo de la enfermedad 4).
Atrofia muscular predominante distal. Miotonía por percusión
La miastenia gravis es el diagnóstico diferencial más importante porque causa ptosis y trastornos del movimiento ocular. La miastenia gravis se caracteriza por variación diurna y fatigabilidad fácil, y se observa mejoría con la prueba de Tensilon o la prueba de la bolsa de hielo. La ptosis en la CPEO no es fluctuante y no mejora con estas pruebas.
No existe un tratamiento curativo establecido para la CPEO. El tratamiento sintomático para mantener la calidad de vida (QOL) es el pilar del tratamiento.
Se pueden probar los siguientes medicamentos para compensar la disfunción mitocondrial.
Coenzima Q10 (Ubidecarenona/Neuquinone®): Asiste la transferencia de electrones en la fosforilación oxidativa. Puede observarse mejoría en algunos casos.
Complejo de vitamina B: Apoya el metabolismo como coenzimas.
Vitamina C: Se usa por sus efectos antioxidantes.
L-carnitina: Asiste el metabolismo de ácidos grasos.
Indicaciones: Cuando la función del elevador está moderadamente conservada o mejor.
Método: Se realiza avance o resección del músculo elevador del párpado superior. Se avanza y fija la aponeurosis mediante abordaje transcutáneo, con o sin plicatura adicional del músculo de Müller.
Nota: En la CPEO, la función del elevador empeora progresivamente, por lo que el efecto puede disminuir a largo plazo.
Suspensión frontal
Indicaciones: Cuando la función del elevador es deficiente (menos de 4 mm). En CPEO, a menudo se elige esta opción.
Método: Conectar el párpado superior al músculo frontal utilizando fascia autóloga (fascia lata, fascia temporal), lámina de Gore-Tex®, hilo de nailon, varilla de silicona, etc.
Es esencial una evaluación preoperatoria cuidadosa por parte de un cirujano oculoplástico experimentado. Si no hay fenómeno de Bell, se debe mantener una corrección insuficiente.
Lentes prismáticos: Efectivos para reducir la diplopía en desviaciones de ángulo pequeño.
Cirugía de estrabismo: Acortamiento del músculo con alteración del movimiento ocular para corregir la posición primaria. Si el ángulo es grande, también se puede realizar un retroceso del músculo antagonista. Debido a la naturaleza progresiva de la enfermedad, es posible la recurrencia.
Evaluación de la función cardíaca: El KSS conlleva riesgo de trastornos de la conducción cardíaca (bloqueo AV), que requieren ECG periódicos y consideración de implantación de marcapasos.
Anomalías endocrinas: Se requiere manejo de diabetes, deficiencia de hormona de crecimiento, baja estatura, etc.
Evaluación auditiva: Se recomiendan pruebas auditivas periódicas para la hipoacusia neurosensorial.
Q¿Puede recurrir la ptosis después de la cirugía?
A
Dado que la CPEO es una enfermedad progresiva, la función del elevador puede empeorar gradualmente después de la cirugía, lo que lleva a la recurrencia de la ptosis. En niños, a menudo es necesaria una reoperación debido al crecimiento. Los materiales utilizados en la suspensión frontal también pueden perder fuerza de tracción a largo plazo. Es importante un seguimiento regular.
La esencia de la CPEO es la alteración de la fosforilación oxidativa debida a mutaciones en el ADNmt o el ADNn.
Las mitocondrias poseen su propio ADN (ADNmt), que codifica 13 proteínas necesarias para la fosforilación oxidativa. Las deleciones o mutaciones puntuales en el ADNmt reducen la actividad de las enzimas de la cadena de transporte de electrones, lo que provoca una producción insuficiente de ATP. Los músculos extraoculares tienen un mayor contenido mitocondrial en comparación con los músculos esqueléticos y tienen mayores demandas metabólicas para mantener la resistencia a la fatiga. Esta característica se considera una de las razones por las que los músculos extraoculares se ven afectados selectivamente en la CPEO.
La heteroplasmia es un concepto patológico importante en la CPEO. Es un fenómeno en el que el ADNmt normal y el mutante coexisten dentro de una misma célula. Cuando la proporción de ADNmt mutante supera un umbral (generalmente más del 80%), las mitocondrias se vuelven disfuncionales.
Visuttijai et al. (2021) informaron dos nuevas mutaciones puntuales en el gen MT-TN (m.5669G>A y m.5702delA). El análisis de fibras musculares individuales mostró que la carga mutacional en las fibras COX negativas promedió el 93%, significativamente diferente de las fibras COX normales (32% y 57%) (P < 0.0001). El umbral para la disfunción de la COX fue superior al 80% en ambos casos 2).
Katayama Ueda et al. (2025) identificaron una nueva mutación en el gen tRNAGlu (m.14677T>C) en un caso de CPEO en un varón japonés. La mediana de la carga mutacional en las fibras rojas rasgadas fue del 88.1%, significativamente mayor que en las fibras no rojas rasgadas (mediana del 17.1%) (P = 0.03) 3).
En cuanto a las mutaciones del ADNn, el gen POLG es una de las causas genéticas nucleares más comunes de CPEO. POLG codifica la ADN polimerasa γ, necesaria para la replicación del ADNmt; sus mutaciones causan deleciones múltiples del ADNmt a través de la alteración de la replicación del ADNmt.
Liu et al. (2023) informaron el caso de una mujer de 38 años con una mutación conocida c.2857C>T (p.R953C) y una nueva mutación c.2391G>C (p.M797I) en el gen POLG. Desarrolló ptosis además de debilidad y entumecimiento en las extremidades, y la biopsia muscular reveló fibras rojas rasgadas 5).
7. Investigación más reciente y perspectivas futuras (informes en fase de investigación)
En la encefalomiopatía mitocondrial de inicio en la infancia, se ha sugerido que la terapia combinada con ácido 5-aminolevulínico (5-ALA) y hierro aumenta la producción de ATP, y se han iniciado ensayos clínicos. Sin embargo, su eficacia en la CPEO es desconocida.
KH176 es un modulador redox dirigido a las mitocondrias que reduce el daño celular mediado por especies reactivas de oxígeno.
Se ha iniciado un ensayo clínico de fase II que evalúa la seguridad y eficacia de KH176 (100 mg dos veces al día) en pacientes con CPEO (NCT04604548)5).
Se están realizando intentos de introducir genes normales utilizando vectores seguros. Además, se ha informado un método para suprimir el mtDNA mutante con ARN oligo utilizando las características de la heteroplasmia.
Como técnicas de manipulación del genoma mitocondrial, se está estudiando la eliminación selectiva del mtDNA mutante mediante enzimas de restricción, TALEN, ZFN y CRISPR1). La terapia de reemplazo en la línea germinal (transferencia pronuclear, transferencia del huso ovocitario) también se encuentra en etapa de modelos animales y ensayos clínicos.
Fan et al. (2021) detectaron un pico doble a 1–2 ppm (que sugiere acumulación de lactato) en la espectroscopia de RM muscular de pacientes con síndrome CPEO-plus. Fue más prominente en músculos edematosos, lo que indica su potencial como biomarcador para el monitoreo de la enfermedad4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.