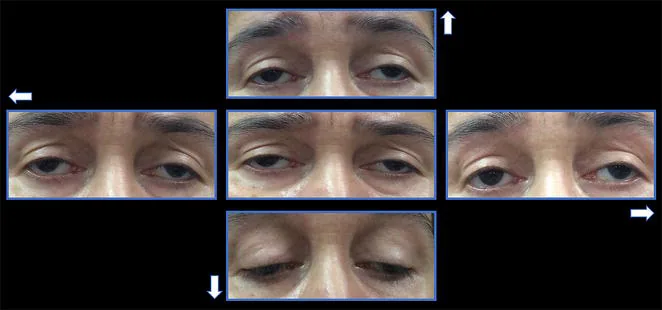
L’ophtalmoplégie externe progressive chronique (CPEO) est un type d’encéphalomyopathie mitochondriale, un groupe de maladies systémiques dues à un dysfonctionnement mitochondrial. Elle affecte sélectivement les muscles extra-oculaires, provoquant un ptosis bilatéral lentement progressif et une limitation des mouvements oculaires.
C’est le phénotype le plus fréquent des maladies mitochondriales. Selon une base de données de cohorte britannique, la prévalence est d’environ 1/30 000 et l’incidence de 1 à 2/100 0001).
L’histoire de la CPEO commence avec la première description par von Graefe en 1868. En 1958, Kearns et Sayre ont décrit la triade CPEO, rétinite pigmentaire et trouble de la conduction cardiaque. En 1972, des fibres rouges déchiquetées (ragged red fibers) ont été découvertes sur biopsie musculaire, et des délétions de l’ADNmt ont été détectées en 1988-1989. En 2000, la première mutation de l’ADN nucléaire (ADNn) associée à des délétions multiples de l’ADNmt a été identifiée1).
Cliniquement, la CPEO est divisée en deux types principaux.
CPEO isolé : seuls les symptômes oculaires sont présents, sans signes systémiques. L’apparition est typique entre 30 et 40 ans.
CPEO-plus : accompagné de symptômes systémiques tels que surdité de perception, dysphagie, troubles de la conduction cardiaque, ataxie cérébelleuse, faiblesse musculaire proximale. Le syndrome de Kearns-Sayre (KSS) en est un exemple représentatif.
Il existe également des cas survenant autour de la puberté, avec peu de signes cliniques systémiques, et les patients consultent souvent d’abord un ophtalmologiste.
QQuelle est la différence entre CPEO et CPEO-plus ?
A
Le CPEO isolé ne présente qu’un ptosis et des troubles oculomoteurs, sans symptômes systémiques. Le CPEO-plus associe des symptômes systémiques tels que surdité de perception, dysphagie, troubles de la conduction cardiaque, ataxie cérébelleuse. Le syndrome de Kearns-Sayre est une maladie représentative du CPEO-plus, survenant avant 20 ans, avec rétinite pigmentaire et troubles de la conduction cardiaque.
Alvaro Ortiz, Juan Arias, Pedro Cárdenas et al. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. International Journal of Retina and Vitreous. 2017 Jul 10; 3:24. Figure 1. PMCID: PMC5502322. License: CC BY.
L’apparition des symptômes du CPEO est extrêmement progressive. Le premier symptôme est souvent un ptosis.
Ptosis : le patient consulte pour une sensation que « la paupière ne se lève pas bien ». Il progresse progressivement d’unilatéral à bilatéral. Il n’est pas rare que le patient ne s’en rende compte qu’après que quelqu’un d’autre le lui ait signalé.
Troubles oculomoteurs : apparaissent quelques années après le ptosis. Une limitation des mouvements dans toutes les directions progresse progressivement.
Absence de diplopie : comme les deux yeux sont atteints symétriquement, les patients ne ressentent souvent pas de diplopie malgré des troubles oculomoteurs marqués.
Position compensatrice de la tête : le patient compense le manque de mouvements oculaires par des changements de position de la tête.
Intolérance à l’effort : dans les cas accompagnés de faiblesse musculaire généralisée, le patient se plaint de fatigabilité.
L’absence de douleur est caractéristique de la CPEO. La douleur oculaire, l’exophtalmie ou les anomalies pupillaires suggèrent d’autres étiologies.
Signes cliniques (observés par le médecin lors de l’examen)
Ptosis : bilatéral, symétrique, avec une fonction du muscle élévateur réduite à moins de 8-10 mm 1). Dans les cas avancés, le ptosis est sévère.
Limitation des mouvements oculaires : dans toutes les directions. Un exotropisme peut être présent en position primaire.
Préservation pupillaire : la CPEO ne s’accompagne pas d’anomalies pupillaires. Une dilatation pupillaire suggère une autre cause, comme une paralysie du nerf oculomoteur.
Fermeture incomplète des paupières : associée au ptosis, due à une faiblesse du muscle orbiculaire, pouvant entraîner une kératite d’exposition.
Signes rétiniens : l’angiographie à la fluorescéine peut montrer une hyperfluorescence granulaire au niveau de l’épithélium pigmentaire rétinien. Une rétinopathie en sel et poivre évoque un syndrome de Kearns-Sayre.
QPourquoi la diplopie est-elle rarement ressentie dans la CPEO ?
A
Dans la CPEO, les deux yeux sont atteints de manière symétrique et progressive, ce qui réduit la différence de mouvement entre les yeux et rend la diplopie rare. De plus, la progression très lente permet une adaptation centrale. Seul un tiers des patients environ présente une diplopie constante ou intermittente.
Les causes de la CPEO se divisent en mutations de l’ADN mitochondrial (ADNmt) et mutations de l’ADN nucléaire (ADNn).
Mutations de l'ADNmt
Grande délétion unique : cause la plus fréquente, représentant 60 à 80 % des cas. La plupart des délétions mesurent entre 1,3 et 1,9 kb. Les cas sporadiques suggèrent une mutation de novo.
Mutations ponctuelles : les gènes d’ARNt sont des points chauds. Au moins 35 mutations ponctuelles peuvent se présenter comme une CPEO isolée. Le gène MT-TN (ARNtAsn) est un point chaud pour la PEO sporadique, et 7 des 11 mutations entraînent un phénotype PEO 2).
Mode de transmission : hérédité maternelle. L’hétéroplasmie (mélange d’ADNmt normal et muté) est caractéristique ; lorsque la proportion de mutation dépasse 80 %, un dysfonctionnement cellulaire devient apparent2)3).
Mutation de l'ADNn
Gènes impliqués dans le maintien de l’ADNmt : des mutations de POLG (ADN polymérase γ), TWNK (Twinkle), SLC25A4 (ANT1), POLG2, RRM2B, RNASEH1, MGME1, DNA2, TK2, DGUOK, entre autres, ont été rapportées1)5).
Délétions multiples de l’ADNmt : les mutations de l’ADNn entraînent des délétions multiples de l’ADNmt via une altération de la réplication et de la réparation de l’ADNmt.
Mode de transmission : autosomique dominant ou autosomique récessif. L’évaluation des antécédents familiaux est importante.
La raison pour laquelle les muscles extra-oculaires sont sélectivement touchés est qu’ils ont une teneur plus élevée en mitochondries et des besoins métaboliques plus importants que les muscles squelettiques, ce qui les rend plus vulnérables aux troubles de la phosphorylation oxydative.
QLa CPEO est-elle héréditaire ?
A
Le mode de transmission varie selon le gène responsable. Les cas sporadiques de mutation de l’ADNmt sont des mutations de novo et le risque génétique est faible. Les cas de mutation de l’ADNn présentent une transmission autosomique dominante ou récessive, avec des cas familiaux. Les mutations de l’ADNmt peuvent également être transmises par hérédité maternelle. Une consultation génétique est recommandée.
Le CPEO repose principalement sur un diagnostic clinique, mais divers examens sont utiles pour confirmer le diagnostic ou pour le diagnostic différentiel. Dans les cliniques spécialisées, l’attention peut se concentrer sur une maladie spécifique, et un ptosis ou des troubles des mouvements oculaires peuvent être négligés 6).
Analyses sanguines : Une augmentation du lactate sérique, de la créatine kinase (CK) et du lactate du liquide céphalorachidien peut être observée, mais la sensibilité et la spécificité ne sont pas élevées. Une augmentation du pyruvate sanguin et du rapport lactate/pyruvate peut également être utile.
Tests d’anticorps : La confirmation de la négativité des anticorps anti-récepteur de l’acétylcholine et des auto-anticorps thyroïdiens est utile pour exclure la myasthénie grave et l’ophtalmopathie thyroïdienne.
Biomarqueurs : Le facteur de croissance des fibroblastes 21 (FGF-21) et le facteur de différenciation de croissance 15 (GDF-15) attirent l’attention en tant que tests de dépistage non invasifs 1).
Coloration de Gomori au trichrome : On observe des fibres rouges déchiquetées (ragged red fibers). Les mitochondries accumulées dans ou autour des fibres musculaires sont fortement colorées.
Double coloration COX/SDH : Des fibres négatives à la cytochrome c oxydase (COX) apparaissent en mosaïque.
Microscopie électronique : Des mitochondries géantes et de morphologie anormale sont observées entre les fibres musculaires.
L’électromyographie des muscles extra-oculaires montre un motif d’interférence avec une amplitude légèrement plus faible que celle des muscles normaux, mais avec une décharge suffisante même en l’absence de mouvements oculaires. Utile pour le diagnostic différentiel avec les maladies neurogènes.
Délétion de l’ADNmt : La méthode de Southern blot sur échantillon de biopsie musculaire squelettique est fiable. La méthode long PCR peut parfois permettre la détection dans le sang périphérique.
Mutation ponctuelle de l’ADNmt : L’analyse complète de l’ADNmt par séquençage de nouvelle génération (NGS) est efficace. Parfois, elle n’est détectable que dans le tissu musculaire et non dans le sang 2)3).
Mutation de l’ADNn : un séquençage de l’exome des gènes associés tels que POLG, TWNK, SLC25A4 est effectué 5).
IRM cérébrale : peut montrer des hypersignaux de la substance blanche, une atrophie corticale, une atrophie cérébelleuse, des hypersignaux du tronc cérébral, etc. Un cas de KSS a rapporté une atrophie cérébelleuse unilatérale progressive dans le temps 7).
Spectroscopie par résonance magnétique (SRM) : peut détecter de manière non invasive l’accumulation de lactate dans les muscles, potentiellement utile pour le suivi de la maladie 4).
Atrophie musculaire prédominant aux muscles distaux. Myotonie de percussion
La myasthénie grave est la maladie différentielle la plus importante car elle provoque un ptosis et des troubles des mouvements oculaires. Dans la myasthénie grave, les fluctuations diurnes et la fatigabilité sont caractéristiques, et une amélioration est observée avec le test au Tensilon ou le test du glaçon. Le ptosis de la CPEO est non fluctuant et ne s’améliore pas avec ces tests.
Il n’existe pas de traitement curatif établi pour la CPEO. Le maintien de la qualité de vie (QdV) par un traitement symptomatique est au cœur de la prise en charge.
Indication : lorsque la fonction du muscle releveur est modérément ou bien préservée.
Méthode : avancement ou résection du muscle releveur de la paupière supérieure. Par voie percutanée, l’aponévrose est avancée et fixée, avec un tucking du muscle de Müller si nécessaire.
Attention : dans la CPEO, la fonction du muscle releveur se détériore progressivement, donc l’effet peut diminuer à long terme.
Suspension au muscle frontal
Indications : lorsque la fonction du muscle releveur est insuffisante (moins de 4 mm). Dans la CPEO, cette option est souvent choisie.
Méthode : connexion de la paupière supérieure au muscle frontal à l’aide de fascia autologue (fascia lata, fascia temporal), feuille de Gore-Tex®, fil de nylon, tige de silicone, etc.
Attention : une hypercorrection peut entraîner un lagophtalmos et une exposition cornéenne, avec risque de kératopathie d’exposition et d’ulcère cornéen.
Une évaluation préopératoire minutieuse par un chirurgien oculoplastique expérimenté est indispensable. En l’absence de réflexe de Bell, il convient de viser une sous-correction.
Lentilles prismatiques : efficaces pour réduire la diplopie en cas de petit angle de déviation oculaire.
Chirurgie du strabisme : raccourcissement du muscle oculomoteur déficient pour corriger la position primaire du regard. En cas d’angle important, une récession du muscle antagoniste peut être associée. En raison de la nature progressive de la maladie, une récidive est possible.
Évaluation cardiaque : dans le KSS, il existe un risque de trouble de la conduction cardiaque (bloc auriculo-ventriculaire) ; une électrocardiographie régulière et la décision de poser un stimulateur cardiaque sont nécessaires.
Anomalies endocriniennes : une prise en charge du diabète, du déficit en hormone de croissance, de la petite taille, etc., est requise.
Évaluation auditive : des examens audiométriques réguliers sont recommandés en cas de surdité de perception.
QLe ptosis peut-il récidiver après une chirurgie ?
A
La CPEO étant une maladie progressive, la fonction du muscle releveur peut se détériorer progressivement après la chirurgie, entraînant une récidive du ptosis. Chez les enfants, une réintervention est souvent nécessaire en raison de la croissance. Les matériaux utilisés pour la suspension frontale peuvent également voir leur force de traction diminuer à long terme. Un suivi régulier est important.
L’essence du CPEO est un trouble de la phosphorylation oxydative dû à des mutations de l’ADNmt ou de l’ADNn.
Les mitochondries possèdent leur propre ADN (ADNmt) qui code 13 protéines nécessaires à la phosphorylation oxydative. Les délétions ou mutations ponctuelles de l’ADNmt réduisent l’activité des enzymes de la chaîne respiratoire, entraînant un déficit en ATP. Les muscles extra-oculaires ont une teneur plus élevée en mitochondries que les muscles squelettiques et ont des besoins métaboliques plus importants pour maintenir leur résistance à la fatigue. Cette caractéristique est considérée comme l’une des raisons pour lesquelles les muscles extra-oculaires sont sélectivement touchés dans le CPEO.
L’hétéroplasmie est un concept physiopathologique important du CPEO. Il s’agit de la coexistence d’ADNmt normal et muté dans une même cellule. Lorsque la proportion d’ADNmt muté dépasse un seuil (généralement >80%), la mitochondrie devient dysfonctionnelle.
Visuttijai et al. (2021) ont rapporté deux nouvelles mutations ponctuelles du gène MT-TN (m.5669G>A et m.5702delA). L’analyse de fibres musculaires uniques a montré que le pourcentage de mutation dans les fibres COX-négatives était en moyenne de 93 %, significativement différent des fibres COX-normales (32 % et 57 %) (P < 0,0001). Le seuil de dysfonctionnement de la COX était supérieur à 80 % dans les deux cas 2).
Katayama Ueda et al. (2025) ont identifié une nouvelle mutation du gène tRNAGlu (m.14677T>C) chez un homme japonais atteint de CPEO. Le pourcentage médian de mutation dans les fibres ragged-red était de 88,1 %, significativement plus élevé que dans les fibres non ragged-red (médiane 17,1 %) (P = 0,03) 3).
En ce qui concerne les mutations de l’ADNn, le gène POLG est l’une des causes nucléaires les plus courantes du CPEO. POLG code l’ADN polymérase γ nécessaire à la réplication de l’ADNmt, et ses mutations entraînent des délétions multiples de l’ADNmt via un trouble de la réplication.
Liu et al. (2023) ont rapporté le cas d’une femme de 38 ans présentant une mutation connue c.2857C>T (p.R953C) et une nouvelle mutation c.2391G>C (p.M797I) du gène POLG. Elle présentait une faiblesse et un engourdissement des membres ainsi qu’un ptosis, et une biopsie musculaire a révélé des fibres ragged-red 5).
7. Recherches récentes et perspectives d’avenir (rapports de stade de recherche)
Dans l’encéphalomyopathie mitochondriale à début infantile, la thérapie combinée d’acide 5-aminolévulinique (5-ALA) et de fer a été suggérée pour augmenter la production d’ATP, et des essais cliniques ont été lancés. Cependant, son efficacité dans le CPEO est inconnue.
KH176 est un agent régulateur redox ciblant les mitochondries, qui réduit les dommages cellulaires médiés par les espèces réactives de l’oxygène.
Un essai clinique de phase II évaluant la sécurité et l’efficacité de KH176 (100 mg deux fois par jour) chez des patients atteints de CPEO a été lancé (NCT04604548)5).
Des tentatives sont en cours pour introduire un gène normal à l’aide d’un vecteur sûr. De plus, une méthode utilisant l’ARN oligo pour supprimer l’ADNmt mutant en exploitant les caractéristiques de l’hétéroplasmie a été rapportée.
En tant que techniques de manipulation du génome mitochondrial, l’élimination sélective de l’ADNmt mutant par des enzymes de restriction, TALEN, ZFN et CRISPR est étudiée1). La thérapie de remplacement au niveau de la lignée germinale (transfert de pronoyau, transfert de fuseau ovocytaire) est également au stade de modèles animaux et d’essais cliniques.
Fan et al. (2021) ont détecté un double pic à 1-2 ppm (suggérant une accumulation de lactate) en spectroscopie par résonance magnétique musculaire chez des patients atteints du syndrome CPEO-plus. Ce pic était plus prononcé dans les muscles œdémateux, indiquant un potentiel biomarqueur pour le suivi de la maladie4).
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain sciences. 2024;14(2). doi:10.3390/brainsci14020135. PMID:38391710; PMCID:PMC10887352.
Visuttijai K, Hedberg-Oldfors C, Lindgren U, Nordström S, Elíasdóttir Ó, Lindberg C, et al. Progressive external ophthalmoplegia associated with novel MT-TN mutations. Acta neurologica Scandinavica. 2021;143(1):103-108. doi:10.1111/ane.13339. PMID:32869280; PMCID:PMC7756270.
Ueda NK, Mimaki M, Ito S, Murakami A, Yokoi S, Nishino I, Katsuno M, Goto YI. A novel m.14677 T > C variant in mitochondrial tRNA(Glu) gene causes chronic progressive external ophthalmoplegia. Journal of human genetics. 2025;70(10):537-540. doi:10.1038/s10038-025-01381-7. PMID:40770229; PMCID:PMC12460166.
Fan SP, Hsueh HW, Huang HC, et al. Lactate peak in muscle disclosed by magnetic resonance spectroscopy in a patient with CPEO-plus syndrome. eNeurologicalSci. 2021;24:100360. doi:10.1016/j.ensci.2021.100360.
Liu H, Gao M, Sun Q, Chen S, Luo Y, Yang H, et al. A case of mitochondrial myopathy and chronic progressive external ophthalmoplegia. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences. 2023;48(11):1760-1768. doi:10.11817/j.issn.1672-7347.2023.220605. PMID:38432868; PMCID:PMC10929950.
Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G, Karampelas M. Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination. Clinical optometry. 2021;13:335-339. doi:10.2147/OPTO.S342972. PMID:34992483; PMCID:PMC8714969.
Zhao H, Shi M, Yang F, Yang X. Kearns-Sayre syndrome with rare imaging finding of SLC25A4 Mutation. Neurosciences (Riyadh, Saudi Arabia). 2022;27(2):111-115. doi:10.17712/nsj.2022.2.20210123. PMID:35477912; PMCID:PMC9257918.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.