Les neuropathies optiques héréditaires se divisent en deux grandes catégories : l’ADOA, due à des mutations de gènes nucléaires et à transmission mendélienne, et la neuropathie optique héréditaire de Leber (LHON), due à des mutations de l’ADN mitochondrial et à transmission maternelle. L’atrophie optique de transmission autosomique récessive inclut le syndrome de Wolfram. L’ADOA et la LHON partagent un mécanisme commun de dégénérescence des cellules ganglionnaires rétiniennes (RGC) due à un dysfonctionnement mitochondrial, mais leurs présentations cliniques et leurs causes génétiques diffèrent6).
QQuelle est la fréquence de l'atrophie optique autosomique dominante ?
A
La prévalence est estimée entre 1:12 000 et 1:50 000, ce qui en fait la neuropathie optique héréditaire la plus fréquente2). Au Royaume-Uni, la prévalence globale des neuropathies optiques héréditaires est d’environ 1:25 0006).
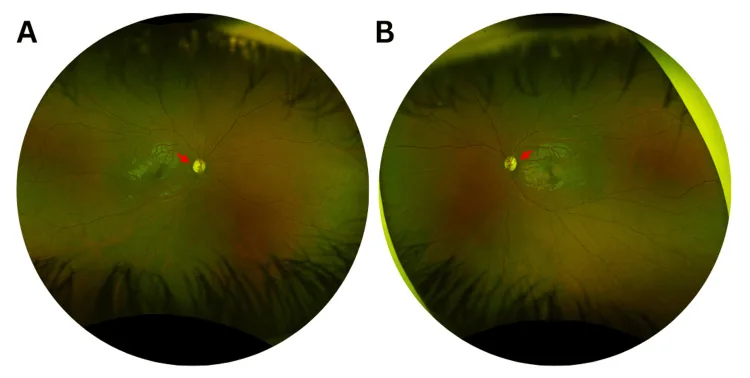
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
Photographies couleur du fond d’œil de l’œil droit (A) et de l’œil gauche (B) lors de la première consultation, montrant une pâleur temporale légère de la papille optique (flèches rouges) et un élargissement de l’excavation papillaire dans les deux yeux. Cela correspond à la pâleur papillaire décrite dans la section « 2. Principaux symptômes et signes cliniques ».
L’apparition typique se situe dans la première ou la deuxième décennie de la vie. En raison de la progression lente, de nombreux patients ne peuvent pas identifier précisément le moment du début.
Baisse de l’acuité visuelle : bilatérale, symétrique, d’évolution lente et insidieuse.
Circonstance de découverte : souvent découverte pendant l’enfance comme un trouble du développement de la vision binoculaire. Les symptômes subjectifs sont rares, et elle peut être découverte fortuitement lors d’un examen de dépistage.
Degré de l’acuité visuelle : plus de 80 % des patients conservent une acuité visuelle d’au moins 20/200 (0,1), mais certains cas présentent une acuité visuelle corrigée inférieure ou égale à 0,1.
Pénétrance : varie de 43 à 100 % selon les familles2). Certains cas conservent une fonction visuelle relativement bonne (meilleure acuité visuelle corrigée [BCVA] 0,6 à 1,0) jusqu’à l’âge mûr2).
Pas de différence de sexe : aucune différence de sexe dans l’apparition.
QL'ADOA est-elle possible même avec une bonne acuité visuelle ?
A
Oui. Dans un rapport de Tachibana et al. (2025), une nouvelle mutation de l’OPA1 a été identifiée chez un patient qui conservait une meilleure acuité visuelle corrigée de 0,8/0,6 à l’âge de 56 ans2). La pénétrance varie de 43 à 100 %, et des anomalies peuvent être détectées par OCT uniquement chez des porteurs asymptomatiques.
Les trois principaux signes sont la papille optique, la vision des couleurs et le champ visuel.
Papille optique : pâleur cunéiforme temporale typique. Une atrophie diffuse peut également être observée.
Dyschromatopsie : peut se présenter comme une tritanopie acquise.
Déficit du champ visuel : scotome central, scotome centro-cæcal et scotome paracentral sont les plus courants. Certains cas présentent peu ou pas d’anomalies.
Tomographie par cohérence optique (OCT) : amincissement des couches internes de la rétine, principalement du faisceau papillo-maculaire. L’amincissement temporal de la couche des fibres nerveuses rétiniennes (RNFL) est caractéristique. Un œdème maculaire microkystique (MME) peut être observé.
Électrophysiologie : Les PEV montrent une diminution d’amplitude et un allongement de latence. L’électrorétinogramme multifocal (ERGmf) montre une réduction de la composante N95.
L’ADOA peut présenter un phénotype DOA plus (DOA plus) avec des anomalies systémiques autres que l’atrophie optique. 20 à 30 % des patients présentent une surdité, une neuropathie périphérique, une myopathie, une ataxie, une ophtalmoplégie externe progressive chronique (CPEO), etc. 3).
Forme typique (DOA)
Symptômes : Baisse progressive et bilatérale de l’acuité visuelle.
Signes papillaires : Pâleur temporale en forme de coin, signe cardinal.
Symptômes généraux : Atrophie optique uniquement.
Acuité visuelle : Plus de 80 % des patients conservent une acuité ≥ 0,1.
DOA plus
Fréquence : Survient chez 20 à 30 % des patients 3).
Complications : Surdité, neuropathie périphérique, myopathie, ataxie, CPEO, etc.
Forme sévère : Les mutations bialléliques de OPA1 entraînent un syndrome de Behr (début précoce, déficience visuelle sévère, ataxie, convulsions) 3).
Plus de 60 % des cas d’ADOA sont dus à des mutations du gène OPA1 6). OPA1 est situé sur le chromosome 3q28-q29 et plus de 500 mutations pathogènes ont été identifiées 6). Au Japon, la mutation c.2708_2711 delTTAG est une mutation fréquente.
Haploinsuffisance : La plupart des mutations OPA1 entraînent une terminaison prématurée de la traduction, réduisant la quantité de protéine OPA1 6).
Pénétration incomplète : complique le diagnostic, le pronostic et le conseil génétique6).
Mutations de novo : surviennent à un taux élevé, donc l’absence d’antécédents familiaux n’exclut pas le diagnostic1).
Dans les cas négatifs pour OPA1, il est nécessaire de rechercher d’autres mutations génétiques. Les principaux gènes responsables sont listés ci-dessous.
Gène
Maladie associée
Remarques
OPA1
ADOA (forme typique, DOA plus)
Plus de 60 % du total6)
AFG3L2
Atrophie optique de type 12 (OAT12), SCA28
Environ 3 % des neuropathies optiques héréditaires1)
OPA3
Atrophie optique dominante avec cataracte et surdité (syndrome de Costeff)
OMIM #258501
WFS1
Syndrome de Wolfram-like
OMIM #222370, #614296
DNM1L
Atrophie optique de type 5 (OPA5)
Régulation de la division mitochondriale
Brodsky et al. (2023) ont identifié une atrophie optique de type 12 due à une mutation c.1064C>T (p.Thr355Met) du gène AFG3L2 chez un père et sa fille d’origine est-africaine (somalienne)1). Dans une étude de cohorte de 2186 cas, AFG3L2 figurait parmi les 10 principaux gènes responsables de la neuropathie optique héréditaire, représentant 14 cas sur 451 (3 %).
QExiste-t-il d'autres gènes responsables en dehors de OPA1 ?
A
Oui. Plusieurs gènes ont été identifiés, notamment AFG3L2, OPA3, WFS1, DNM1L (OPA5). En particulier, AFG3L2 représente environ 3 % des neuropathies optiques héréditaires1), et si OPA1 est négatif, une recherche exhaustive par séquençage de l’exome ou du génome est importante.
En cas de trouble du développement visuel bilatéral d’origine inconnue découvert à l’âge scolaire, suspecter cette maladie. Des antécédents familiaux de symptômes similaires sont un indice important, mais en raison d’une pénétrance incomplète, il peut n’y avoir aucun antécédent familial.
Test de Farnsworth-Munsell 100 hue : montre un axe de tritanopie.
Périmétrie automatique bleu-jaune : détecte plus facilement une baisse de sensibilité que le champ visuel blanc standard2).
OCT : évalue un amincissement de la RNFL prédominant dans les quadrants temporal et inférieur. Des anomalies peuvent être détectées par OCT seul chez les porteurs asymptomatiques.
Dans le cas d’un homme de 56 ans rapporté par Tachibana et al. (2025), le champ visuel blanc HFA 24-2 était normal, mais une baisse de sensibilité a été détectée au champ visuel bleu-jaune2). L’OCT a montré un amincissement temporal de la RNFL, et la CFF était réduite à 30/31 Hz (normal >39 Hz).
Test génétique OPA1 : nécessaire pour un diagnostic définitif. Les tests externes ne sont pas encore répandus et nécessitent une référence à un centre spécialisé.
Séquençage de l’exome/génome : chez les patients OPA1-négatifs, la recherche d’autres gènes, dont AFG3L2, est importante. Une réanalyse peut parfois conduire à un nouveau diagnostic1).
Tests électrophysiologiques : VEP (amplitude réduite, latence prolongée), pERG (diminution de N95), diminution de la CFF.
OCTA : utile pour évaluer les changements neurovasculaires maculaires et péripapillaires.
Surveillance régulière : recommandée une fois par an. Évaluer l’acuité visuelle, le champ visuel, la vision des couleurs, les muscles oculaires externes et l’audition.
Correction réfractive : correction optimale des erreurs de réfraction et gestion de l’observance du port de lunettes.
Réadaptation visuelle : utiliser des aides visuelles telles que les loupes et les synthèses vocales (text-to-speech) 3).
Les éléments suivants ont été proposés pour réduire le stress oxydatif, mais aucun n’est établi comme traitement standard.
Idébénone : analogue synthétique de la coenzyme Q10 (CoQ10). Elle améliore la respiration mitochondriale en contournant le complexe I de la chaîne de transport d’électrons 6). Des rapports suggèrent qu’elle pourrait stabiliser ou améliorer l’acuité visuelle chez les patients atteints d’ADOA avec mutation OPA1 4)5).
CoQ10, vitamines B12, C, lutéine : proposés comme compléments antioxydants.
Il est important d’expliquer la transmission autosomique dominante et de fournir des informations sur le risque de forme sévère (syndrome de Behr) en cas de mutation biallélique 3).
La baisse de l’acuité visuelle est généralement lentement progressive et suit un cours plus bénin que dans la LHON. Plus de 80 % des patients conservent une acuité visuelle corrigée d’au moins 0,1, mais certains cas peuvent descendre en dessous de 0,1. En raison de l’apparition lente, la prise de conscience peut être tardive et la découverte peut être fortuite lors d’un examen de routine. En l’absence de traitement établi, une surveillance régulière et des soins de basse vision sont importants pour la prise en charge à long terme.
QExiste-t-il un traitement efficace pour l'ADOA ?
A
Il n’existe actuellement aucun traitement efficace établi. Les soins de basse vision constituent le principal traitement. L’idébénone a montré un potentiel de stabilisation ou d’amélioration de l’acuité visuelle dans certaines études4)5), mais n’est pas établi comme traitement standard. Pour les approches thérapeutiques en phase de recherche, voir la section « Recherches récentes et perspectives futures ».
OPA1 est une GTPase de type dynamine située dans la membrane interne mitochondriale. Synthétisée dans le noyau, elle est transportée vers les mitochondries où elle assure les fonctions suivantes6).
Fusion de la membrane interne : maintien du réseau mitochondrial
Maintien de la structure des crêtes : stabilisation des complexes de la chaîne respiratoire
Composition des complexes de transport d’électrons : efficacité de la phosphorylation oxydative
Le principal mécanisme pathologique est l’haploinsuffisance. La plupart des mutations d’OPA1 entraînent une terminaison prématurée de la traduction, réduisant la quantité de protéine OPA16).
Diminution de la protéine OPA1 → augmentation de la fragmentation mitochondriale et du recyclage3) → dysfonctionnement métabolique mitochondrial et altération de la phosphorylation oxydative → augmentation des espèces réactives de l’oxygène (ROS) → apoptose des CGR.
Les CGR du faisceau papillo-maculaire dégénèrent principalement, se manifestant par une atrophie du nerf optique temporal.
AFG3L2 code une sous-unité de la métalloprotéase AAA de la matrice mitochondriale (m-AAA)1). Elle forme un complexe avec SPG7 (paraplegine) et assure le traitement, la maturation et le contrôle qualité des protéines mitochondriales de manière ATP-dépendante. Les mutations entraînent une dégénérescence des CGR similaire à celle d’OPA11).
7. Recherches récentes et perspectives d’avenir (rapports de phase de recherche)
Cible : Exon induisant la dégradation NMD (non-sense mediated decay) du pré-ARNm OPA1.
Mécanisme : Inhibition de l’incorporation de l’exon induisant la NMD, approche indépendante de la mutation pour augmenter la traduction de l’OPA1 sauvage6).
État actuel : Augmentation de la protéine OPA1 et amélioration de la bioénergétique mitochondriale confirmées dans trois lignées cellulaires de patients ADOA6). Nécessite des injections intravitréennes répétées régulières, avec des risques d’endophtalmie et d’uvéite chronique comme défis.
Thérapie génique
Modèle murin : La thérapie génique OPA1 prévient la perte de CGR dans un modèle murin de DOA7).
Optimisation des isoformes : Les isoformes optimisées OPA1 1 et 7 montrent un effet thérapeutique dans un modèle de dysfonctionnement mitochondrial8).
État actuel : Stade préclinique.
Édition génique
CRISPR-Cas9 : La correction de la mutation OPA1 c.1334G>A: p.R445H dans des iPSC restaure l’homéostasie mitochondriale9).
Trans-épissage : Une approche de correction des mutations pathogènes au niveau de l’ARNm est également en cours de recherche6).
Cellules souches : La régénération du nerf optique par des RGC dérivées d’iPSC est à l’étude en phase préclinique6).
La réanalyse itérative du séquençage de l’exome constitue également une avancée diagnostique importante. Avec l’expansion des connaissances génétiques, de nouveaux diagnostics peuvent être obtenus à partir de données auparavant négatives1).
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.