Le neuropatie ottiche ereditarie si dividono in due grandi categorie: l’ADOA, causata da mutazioni di geni nucleari e con ereditarietà mendeliana, e la neuropatia ottica ereditaria di Leber (LHON), causata da mutazioni del DNA mitocondriale e con ereditarietà materna. L’atrofia ottica autosomica recessiva include la sindrome di Wolfram. L’ADOA e la LHON condividono un meccanismo patogenetico comune di degenerazione delle cellule gangliari retiniche (RGC) dovuta a disfunzione mitocondriale, ma differiscono per quadro clinico e cause genetiche6).
QQuanto è frequente l'atrofia ottica autosomica dominante?
A
La prevalenza è stimata tra 1:12.000 e 1:50.000, rendendola la neuropatia ottica ereditaria più comune2). Nel Regno Unito, la prevalenza complessiva di tutte le neuropatie ottiche ereditarie è di circa 1:25.0006).
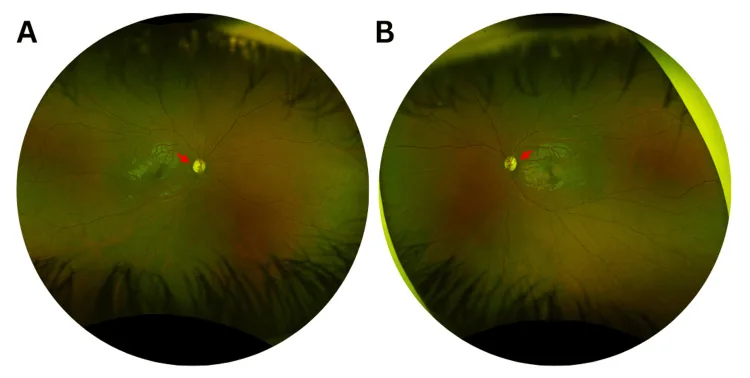
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
Fotografie a colori del fundus dell’occhio destro (A) e sinistro (B) alla prima visita, che mostrano un lieve pallore temporale della papilla ottica (frecce rosse) e un allargamento dell’escavazione in entrambi gli occhi. Ciò corrisponde al pallore papillare discusso nella sezione «2. Principali sintomi e segni clinici».
L’esordio tipico avviene nella prima o seconda decade di vita. A causa della progressione lenta, molti pazienti non riescono a identificare con precisione il momento dell’esordio.
Riduzione dell’acuità visiva: bilaterale, simmetrica, con decorso lento e insidioso.
Circostanza di scoperta: spesso scoperta in età scolare come disturbo dello sviluppo della visione binoculare. I sintomi soggettivi sono scarsi, a volte scoperta casuale durante uno screening.
Grado di acuità visiva: oltre l’80% dei pazienti mantiene un’acuità visiva di almeno 20/200 (0,1), ma in alcuni casi l’acuità visiva corretta è pari o inferiore a 0,1.
Penetranza: varia dal 43 al 100% a seconda della famiglia2). Alcuni casi mantengono una funzione visiva relativamente buona (migliore acuità visiva corretta [BCVA] 0,6–1,0) fino all’età media2).
Nessuna differenza di sesso: non si osserva differenza di sesso nell’insorgenza.
QL'ADOA è possibile anche con una buona acuità visiva?
A
Sì. In un rapporto di Tachibana et al. (2025), è stata identificata una nuova mutazione di OPA1 in un caso che manteneva una migliore acuità visiva corretta di 0,8/0,6 all’età di 56 anni2). La penetranza varia dal 43 al 100% e nei portatori asintomatici possono essere rilevate anomalie solo con OCT.
I tre reperti principali sono papilla ottica, visione dei colori e campo visivo.
Papilla ottica: tipico pallore temporale a forma di cuneo (temporal pallor). Può essere presente anche atrofia diffusa.
Anomalia della visione dei colori: può presentarsi come tritanopia acquisita.
Difetti del campo visivo: scotoma centrale, scotoma centro-cecale e scotoma paracentrale sono i più comuni. In alcuni casi non si riscontrano quasi anomalie.
Campo visivo blu-giallo: più sensibile nel rilevare riduzioni di sensibilità rispetto alla perimetria automatizzata standard con stimolo bianco2).
Tomografia a coerenza ottica (OCT): assottigliamento degli strati interni della retina, principalmente del fascio papillomaculare. Caratteristico l’assottigliamento temporale dello strato delle fibre nervose retiniche (RNFL). Può essere presente edema maculare microcistico (MME).
Elettrofisiologia: I VEP mostrano una riduzione dell’ampiezza e un allungamento della latenza. L’elettroretinogramma pattern (pERG) mostra una riduzione della componente N95.
L’ADOA può presentare un fenotipo DOA plus (DOA plus) con anomalie sistemiche oltre all’atrofia ottica. Il 20-30% dei pazienti presenta comorbidità come sordità, neuropatia periferica, miopatia, atassia, oftalmoplegia esterna progressiva cronica (CPEO), ecc. 3).
Oltre il 60% dei casi di ADOA è causato da mutazioni del gene OPA1 6). OPA1 si trova sul cromosoma 3q28-q29 e sono state identificate oltre 500 mutazioni patogene 6). In Giappone, la mutazione c.2708_2711 delTTAG è nota come mutazione frequente.
Aploinsufficienza: La maggior parte delle mutazioni OPA1 causa una terminazione prematura della traduzione, riducendo la quantità di proteina OPA1 6).
Nei casi negativi per OPA1 è necessario cercare altre mutazioni genetiche. I principali geni causali sono elencati di seguito.
Gene
Malattia associata
Note
OPA1
ADOA (forma tipica, DOA plus)
Oltre il 60% del totale6)
AFG3L2
Atrofia ottica tipo 12 (OAT12), SCA28
Circa il 3% delle neuropatie ottiche ereditarie1)
OPA3
Atrofia ottica dominante + cataratta/sordità (sindrome di Costeff)
OMIM #258501
WFS1
Sindrome di Wolfram-like
OMIM #222370, #614296
DNM1L
Atrofia ottica tipo 5 (OPA5)
Regolazione della divisione mitocondriale
Brodsky et al. (2023) hanno identificato un’atrofia ottica di tipo 12 dovuta a una mutazione c.1064C>T (p.Thr355Met) del gene AFG3L2 in un padre e una figlia di origine est-africana (somala)1). In uno studio di coorte su 2186 casi, AFG3L2 era tra i primi 10 geni causali della neuropatia ottica ereditaria, rappresentando 14 casi su 451 (3%).
QEsistono altri geni causali oltre a OPA1?
A
Sì. Sono stati identificati diversi geni, tra cui AFG3L2, OPA3, WFS1, DNM1L (OPA5). In particolare, AFG3L2 rappresenta circa il 3% delle neuropatie ottiche ereditarie1) e, se OPA1 è negativo, è importante una ricerca esaustiva mediante sequenziamento dell’esoma/genoma.
In caso di disturbo dello sviluppo visivo bilaterale di causa sconosciuta scoperto in età scolare, sospettare questa malattia. Una storia familiare di sintomi simili è un indizio importante, ma a causa della penetranza incompleta potrebbe non esserci storia familiare.
Test di Farnsworth-Munsell 100 hue: mostra un asse di tritanopia.
Perimetria automatica blu-su-giallo: rileva più facilmente una riduzione di sensibilità rispetto al campo visivo bianco standard2).
OCT: valuta un assottigliamento dello RNFL predominante nei quadranti temporale e inferiore. Anche nei portatori asintomatici, le anomalie possono essere rilevate solo con OCT.
Nel caso di un uomo di 56 anni riportato da Tachibana et al. (2025), il campo visivo bianco HFA 24-2 era normale, ma il campo visivo blu-su-giallo ha rilevato una riduzione di sensibilità2). L’OCT ha mostrato un assottigliamento temporale dello RNFL e la CFF era ridotta a 30/31 Hz (normale >39 Hz).
Test genetico OPA1: necessario per la diagnosi definitiva. I test esterni non sono ancora diffusi e richiedono l’invio a un centro specializzato.
Sequenziamento dell’esoma/genoma: nei casi OPA1-negativi, è importante la ricerca di altri geni, incluso AFG3L2. Una rianalisi può talvolta portare a una nuova diagnosi1).
Test elettrofisiologici: VEP (riduzione dell’ampiezza, aumento della latenza), pERG (riduzione di N95), riduzione della CFF.
OCTA: utile per valutare i cambiamenti neurovascolari maculari e peripapillari.
I seguenti sono stati proposti per ridurre lo stress ossidativo, ma nessuno è stabilito come trattamento standard.
Idebenone: analogo sintetico del coenzima Q10 (CoQ10). Migliora la respirazione mitocondriale bypassando il complesso I della catena di trasporto degli elettroni 6). Esistono segnalazioni di possibile stabilizzazione o miglioramento dell’acuità visiva in pazienti con ADOA con mutazione OPA1 4)5).
CoQ10, vitamine B12, C, luteina: proposti come integratori antiossidanti.
È importante spiegare l’ereditarietà autosomica dominante e fornire informazioni sul rischio di forma grave (sindrome di Behr) in caso di mutazione biallelica 3).
La riduzione dell’acuità visiva è generalmente lentamente progressiva e ha un decorso più lieve rispetto alla LHON. Oltre l’80% dei pazienti mantiene un’acuità visiva corretta di 0,1 o superiore, ma in alcuni casi può scendere al di sotto di 0,1. A causa dell’esordio lento, la consapevolezza può essere tardiva e la scoperta può essere accidentale durante gli screening. In assenza di una terapia consolidata, il monitoraggio regolare e le cure per la bassa visione sono importanti per la gestione a lungo termine.
QEsiste un trattamento efficace per l'ADOA?
A
Attualmente non esiste una terapia efficace consolidata. La cura principale è la riabilitazione visiva per ipovedenti (low vision care). L’idebenone ha mostrato potenziale di stabilizzazione o miglioramento dell’acuità visiva in alcuni studi4)5), ma non è stabilito come trattamento standard. Per gli approcci terapeutici in fase di ricerca, vedere la sezione «Ricerche recenti e prospettive future».
OPA1 è una GTPasi correlata alla dinamina situata nella membrana mitocondriale interna. Sintetizzata nel nucleo, viene trasportata nei mitocondri dove svolge le seguenti funzioni6).
Fusione della membrana interna: mantenimento della rete mitocondriale
Mantenimento della struttura delle creste: stabilizzazione dei complessi della catena respiratoria
Costituzione dei complessi di trasporto degli elettroni: efficienza della fosforilazione ossidativa
Il principale meccanismo patologico è l’aploinsufficienza. La maggior parte delle mutazioni di OPA1 causa una terminazione prematura della traduzione, riducendo la quantità di proteina OPA16).
Riduzione della proteina OPA1 → aumento della frammentazione mitocondriale e del riciclaggio3) → alterazione del metabolismo mitocondriale e della fosforilazione ossidativa → aumento delle specie reattive dell’ossigeno (ROS) → apoptosi delle CGR.
Principalmente degenerano le CGR del fascio papillomaculare, manifestandosi come atrofia del nervo ottico temporale.
AFG3L2 codifica una subunità della metalloproteasi AAA della matrice mitocondriale (m-AAA)1). Forma un complesso con SPG7 (paraplegina) e svolge la lavorazione, maturazione e controllo qualità delle proteine mitocondriali in modo ATP-dipendente. Le mutazioni causano degenerazione delle CGR simile a OPA11).
7. Ricerche recenti e prospettive future (rapporti di fase di ricerca)
Bersaglio : Esone che induce la NMD (degradazione mediata da nonsenso) del pre-mRNA di OPA1.
Meccanismo : Inibizione dell’incorporazione dell’esone che induce la NMD, approccio indipendente dalla mutazione per aumentare la traduzione di OPA1 wild-type6).
Stato attuale : Aumento della proteina OPA1 e miglioramento della bioenergetica mitocondriale confermati in tre linee cellulari di pazienti ADOA6). Richiede iniezioni intravitreali ripetute regolari, con rischi di endoftalmite e uveite cronica come sfide.
Terapia genica
Modello murino : La terapia genica OPA1 previene la perdita di RGC in un modello murino di DOA7).
Ottimizzazione delle isoforme : Le isoforme ottimizzate OPA1 1 e 7 mostrano effetto terapeutico in un modello di disfunzione mitocondriale8).
Stato attuale : Fase preclinica.
Editing genetico
CRISPR-Cas9 : La correzione della mutazione OPA1 c.1334G>A: p.R445H in iPSC ripristina l’omeostasi mitocondriale9).
Trans-splicing : Un approccio di correzione delle mutazioni patogene a livello di mRNA è anche in fase di ricerca6).
Cellule staminali: La rigenerazione del nervo ottico mediante RGC derivate da iPSC è in fase di studio preclinico6).
La rianalisi iterativa del sequenziamento dell’esoma è anch’essa un importante progresso diagnostico. Con l’espansione delle conoscenze genetiche, nuovi dati diagnostici possono essere ottenuti da dati precedentemente negativi1).
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.