Die autosomal-dominante Optikusatrophie (ADOA) ist eine erbliche Optikusneuropathie, die durch eine beidseitige progressive Optikusatrophie gekennzeichnet ist. Die OMIM-Nummer ist 165500. Es ist die häufigste erbliche Optikusneuropathie mit einer geschätzten Prävalenz von 1:12.000 bis 1:50.0002).
Erbliche Optikusneuropathien werden grob in zwei Kategorien eingeteilt: ADOA, verursacht durch Kern-Genmutationen und mendelschen Erbgang, und die Lebersche hereditäre Optikusneuropathie (LHON), verursacht durch mitochondriale DNA-Mutationen und mütterliche Vererbung. Die autosomal-rezessive Optikusatrophie umfasst das Wolfram-Syndrom. ADOA und LHON teilen eine gemeinsame Pathologie der Degeneration retinaler Ganglienzellen (RGC) aufgrund mitochondrialer Dysfunktion, unterscheiden sich jedoch im klinischen Bild und den genetischen Ursachen6).
QWie häufig ist die autosomal-dominante Optikusatrophie?
A
Die Prävalenz wird auf 1:12.000 bis 1:50.000 geschätzt, was sie zur häufigsten erblichen Optikusneuropathie macht2). Im Vereinigten Königreich wird die Gesamtprävalenz aller erblichen Optikusneuropathien mit etwa 1:25.000 angegeben6).
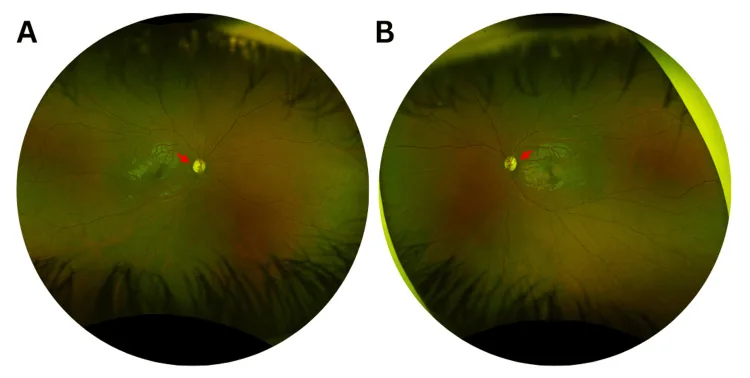
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
Farbfundusfotografien des rechten (A) und linken (B) Auges bei der Erstvorstellung, die eine leichte temporale Papillenabblassung (rote Pfeile) und eine Vergrößerung der Exkavation in beiden Augen zeigen. Dies entspricht der Papillenabblassung, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Der typische Beginn liegt im ersten bis zweiten Lebensjahrzehnt. Aufgrund des langsamen Fortschreitens können viele Patienten den genauen Zeitpunkt des Beginns nicht bestimmen.
Sehverschlechterung: beidseitig, symmetrisch, langsam und schleichend verlaufend.
Entdeckungsanlass: Wird oft im Schulalter als Entwicklungsstörung des binokularen Sehens entdeckt. Subjektive Symptome sind selten, manchmal Zufallsbefund bei Vorsorgeuntersuchungen.
Sehschärfegrad: Über 80 % der Patienten behalten eine Sehschärfe von mindestens 20/200 (0,1), aber in einigen Fällen liegt die korrigierte Sehschärfe bei 0,1 oder darunter.
Penetranz: Je nach Familie zwischen 43 und 100 % variierend2). Einige Fälle behalten bis ins mittlere Alter eine relativ gute Sehfunktion (beste korrigierte Sehschärfe [BCVA] 0,6–1,0)2).
Kein Geschlechtsunterschied: Es besteht kein Geschlechtsunterschied beim Auftreten.
QIst ADOA auch bei guter Sehschärfe möglich?
A
Ja. In einem Bericht von Tachibana et al. (2025) wurde bei einem Fall, der im Alter von 56 Jahren eine beste korrigierte Sehschärfe von 0,8/0,6 aufwies, eine neue Mutation im OPA1-Gen identifiziert2). Die Penetranz variiert zwischen 43 und 100 %, und bei asymptomatischen Trägern können allein mittels OCT Auffälligkeiten festgestellt werden.
Die drei Hauptbefunde sind Sehnervenkopf, Farbsehen und Gesichtsfeld.
Sehnervenkopf: Typisch ist eine keilförmige temporale Abblassung (temporal pallor). Auch diffuse Atrophie kann vorkommen.
Farbsehstörung: Kann als erworbene Tritanopie auftreten.
Gesichtsfeldausfälle: Zentralskotom, zentrozökales Skotom und parazentrales Skotom sind am häufigsten. In manchen Fällen gibt es kaum Auffälligkeiten.
Blau-Gelb-Gesichtsfeld: Empfindlicher für den Nachweis von Empfindlichkeitsminderungen als die standardmäßige automatisierte Perimetrie mit weißem Stimulus2).
Optische Kohärenztomographie (OCT): Verdünnung der inneren Netzhautschichten, hauptsächlich des papillomakulären Bündels. Charakteristisch ist die temporale Verdünnung der retinalen Nervenfaserschicht (RNFL). Ein mikrozystisches Makulaödem (MME) kann auftreten.
Elektrophysiologie: VEP zeigt verminderte Amplitude und verlängerte Latenz. Das Muster-Elektroretinogramm (PERG) zeigt eine reduzierte N95-Komponente.
ADOA kann einen DOA-plus-Phänotyp (DOA plus) mit systemischen Anomalien zusätzlich zur Optikusatrophie aufweisen. 20–30 % der Patienten haben begleitende Hörverlust, periphere Neuropathie, Myopathie, Ataxie, chronisch progressive externe Ophthalmoplegie (CPEO) usw. 3).
Über 60 % der ADOA-Fälle werden durch Mutationen im OPA1-Gen verursacht 6). OPA1 liegt auf Chromosom 3q28-q29, und es wurden über 500 pathogene Mutationen identifiziert 6). In Japan ist c.2708_2711 delTTAG als häufige Mutation bekannt.
Haploinsuffizienz: Die meisten OPA1-Mutationen führen zu einem vorzeitigen Translationsabbruch, wodurch die Menge des OPA1-Proteins reduziert wird 6).
Unvollständige Penetranz: erschwert Diagnose, Prognose und genetische Beratung6).
De-novo-Mutationen: treten mit hoher Rate auf, daher kann eine fehlende Familienanamnese die Diagnose nicht ausschließen1).
Bei OPA1-negativen Fällen müssen andere Genmutationen gesucht werden. Die wichtigsten ursächlichen Gene sind unten aufgeführt.
Gen
Assoziierte Erkrankung
Anmerkungen
OPA1
ADOA (typische Form, DOA plus)
Über 60 % der Gesamtzahl6)
AFG3L2
Optikusatrophie Typ 12 (OAT12), SCA28
Etwa 3 % der hereditären Optikusneuropathien1)
OPA3
Dominante Optikusatrophie mit Katarakt und Schwerhörigkeit (Costeff-Syndrom)
OMIM #258501
WFS1
Wolfram-Syndrom-ähnlich
OMIM #222370, #614296
DNM1L
Optikusatrophie Typ 5 (OPA5)
Regulation der mitochondrialen Teilung
Brodsky et al. (2023) identifizierten eine Optikusatrophie Typ 12 aufgrund einer AFG3L2-Genmutation c.1064C>T (p.Thr355Met) bei einem Vater und seiner Tochter ostafrikanischer (somalischer) Abstammung1). In einer Kohortenstudie mit 2186 Fällen gehörte AFG3L2 zu den Top-10-Genen für hereditäre Optikusneuropathie und machte 14 von 451 Fällen (3 %) aus.
QGibt es außer OPA1 weitere ursächliche Gene?
A
Ja. Mehrere Gene wurden identifiziert, darunter AFG3L2, OPA3, WFS1, DNM1L (OPA5). Insbesondere AFG3L2 macht etwa 3 % der hereditären Optikusneuropathien aus1), und bei negativem OPA1 ist eine umfassende Suche mittels Exom-/Genomsequenzierung wichtig.
Bei einer im Schulalter entdeckten beidseitigen Sehentwicklungsstörung unklarer Ursache sollte an diese Erkrankung gedacht werden. Eine Familienanamnese mit ähnlichen Symptomen ist ein wichtiger Hinweis, kann aber aufgrund unvollständiger Penetranz fehlen.
Farnsworth-Munsell 100 hue Test: zeigt eine Tritanopie-Achse.
Blau-Gelb automatische Gesichtsfelduntersuchung: erkennt leichter eine Empfindlichkeitsminderung als das normale weiße Gesichtsfeld2).
OCT: beurteilt eine RNFL-Ausdünnung mit Betonung der temporalen und unteren Quadranten. Auch bei asymptomatischen Trägern können allein mit OCT Auffälligkeiten festgestellt werden.
Im Fall eines 56-jährigen Mannes von Tachibana et al. (2025) war das HFA 24-2 weiße Gesichtsfeld normal, aber das Blau-Gelb-Gesichtsfeld zeigte eine Empfindlichkeitsminderung2). Das OCT zeigte eine temporale RNFL-Ausdünnung, und die CFF war auf 30/31 Hz (normal >39 Hz) reduziert.
OPA1-Gentest: für die definitive Diagnose erforderlich. Externe Tests sind noch nicht weit verbreitet und erfordern eine Überweisung an ein Zentrum.
Exom-/Genomsequenzierung: bei OPA1-negativen Fällen ist die Suche nach anderen Genen, einschließlich AFG3L2, wichtig. Eine erneute Analyse kann manchmal zu einer neuen Diagnose führen1).
Zur Reduzierung von oxidativem Stress wurden folgende Mittel vorgeschlagen, aber keines ist als Standardtherapie etabliert.
Idebenon: Synthetisches Analogon von Coenzym Q10 (CoQ10). Es verbessert die mitochondriale Atmung durch Umgehung des Komplexes I der Elektronentransportkette 6). Es gibt Berichte über eine mögliche Stabilisierung oder Verbesserung der Sehschärfe bei OPA1-Mutations-ADOA-Patienten 4)5).
CoQ10, Vitamin B12, C, Lutein: Als antioxidative Nahrungsergänzungsmittel vorgeschlagen.
Die Aufklärung über den autosomal-dominanten Erbgang und die Information über das Risiko einer schweren Verlaufsform (Behr-Syndrom) bei biallelischer Mutation ist wichtig 3).
Die Sehverschlechterung verläuft in der Regel langsam fortschreitend und milder als bei LHON. Über 80 % der Patienten behalten eine korrigierte Sehschärfe von 0,1 oder besser, aber es gibt auch Fälle mit einem Abfall unter 0,1. Aufgrund des schleichenden Beginns wird die Erkrankung oft spät bemerkt oder zufällig bei Vorsorgeuntersuchungen entdeckt. Da es keine etablierte Therapie gibt, sind regelmäßige Überwachung und Low-Vision-Versorgung für die Langzeitbehandlung wichtig.
QGibt es eine wirksame Behandlung für ADOA?
A
Derzeit gibt es keine etablierte wirksame Behandlung. Die Sehbehindertenversorgung (Low-Vision-Care) steht im Vordergrund. Idebenon hat in einigen Studien Potenzial zur Stabilisierung oder Verbesserung der Sehkraft gezeigt4)5), ist aber nicht als Standardtherapie etabliert. Für experimentelle Therapieansätze siehe den Abschnitt „Aktuelle Forschung und zukünftige Perspektiven“.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
OPA1 ist eine Dynamin-verwandte GTPase in der inneren Mitochondrienmembran. Es wird im Zellkern synthetisiert, zu den Mitochondrien transportiert und erfüllt folgende Funktionen6).
Fusion der inneren Membran: Aufrechterhaltung des mitochondrialen Netzwerks
Erhaltung der Cristae-Struktur: Stabilisierung der Atmungskettenkomplexe
Aufbau der Elektronentransportkomplexe: Effizienz der oxidativen Phosphorylierung
Der wichtigste pathologische Mechanismus ist die Haploinsuffizienz. Die meisten OPA1-Mutationen führen zu einem vorzeitigen Translationsabbruch, wodurch die Menge des OPA1-Proteins reduziert wird6).
Abnahme des OPA1-Proteins → verstärkte mitochondriale Fragmentierung und erhöhtes Recycling3) → mitochondriale Stoffwechselstörung und Beeinträchtigung der oxidativen Phosphorylierung → Anstieg reaktiver Sauerstoffspezies (ROS) → Apoptose der RGC.
Hauptsächlich degenerieren die RGC des papillomakulären Bündels, was sich als temporale Optikusatrophie äußert.
AFG3L2 kodiert eine Untereinheit der mitochondrialen Matrix-AAA-Metalloprotease (m-AAA)1). Es bildet einen Komplex mit SPG7 (Paraplegin) und führt die Prozessierung, Reifung und Qualitätskontrolle mitochondrialer Proteine ATP-abhängig durch. Mutationen verursachen ähnlich wie bei OPA1 eine RGC-Degeneration1).
7. Aktuelle Forschung und Zukunftsperspektiven (Forschungsstadium-Berichte)
Ziel : Exon, das den NMD (Nonsense-vermittelter Abbau) der OPA1-Prä-mRNA induziert.
Mechanismus : Hemmung des Einbaus des NMD-induzierenden Exons, mutationsunabhängiger Ansatz zur Steigerung der Wildtyp-OPA1-Translation6).
Aktueller Stand : Erhöhte OPA1-Proteinproduktion und verbesserte mitochondriale Bioenergetik in drei ADOA-Patientenzelllinien bestätigt6). Erfordert regelmäßige intravitreale Injektionen, mit Risiken wie Endophthalmitis und chronischer Uveitis als Herausforderung.
Gentherapie
Mausmodell : OPA1-Gentherapie verhindert RGC-Verlust im DOA-Mausmodell7).
Isoform-Optimierung : Optimierte OPA1-Isoformen 1 und 7 zeigen therapeutische Wirkung in einem mitochondrialen Dysfunktionsmodell8).
Aktueller Stand : Präklinische Phase.
Gen-Editierung
CRISPR-Cas9 : Korrektur der OPA1-Mutation c.1334G>A: p.R445H in iPSC stellt die mitochondriale Homöostase wieder her9).
Trans-Spleißen : Ein Ansatz zur Korrektur pathogener Mutationen auf mRNA-Ebene wird ebenfalls erforscht6).
Stammzellen: Die Regeneration des Sehnervs durch iPSC-abgeleitete RGC wird in präklinischen Studien untersucht6).
Die iterative Neuauswertung der Exomsequenzierung ist ebenfalls ein wichtiger diagnostischer Fortschritt. Mit der Erweiterung des genetischen Wissens können aus zuvor negativen Daten neue Diagnosen gewonnen werden1).
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.