نوع کلاسیک (DOA)
علائم: کاهش تدریجی و دوطرفه بینایی.
یافتههای پاپیلاری: رنگپریدگی گوهای گیجگاهی یافته اصلی است.
علائم سیستمیک: فقط آتروفی عصب بینایی.
حدت بینایی: بیش از ۸۰٪ بیماران حدت بینایی ۰.۱ یا بهتر را حفظ میکنند.

آتروفی بینایی اتوزومال غالب (ADOA) یک نوروپاتی بینایی ارثی است که با آتروفی پیشرونده دوطرفه عصب بینایی مشخص میشود. شماره OMIM آن 165500 است. این شایعترین نوروپاتی بینایی ارثی است و شیوع آن 1:12000 تا 1:50000 تخمین زده میشود2).
نوروپاتیهای بینایی ارثی به دو دسته اصلی تقسیم میشوند: ADOA با وراثت مندلی و ژنهای هستهای، و نوروپاتی بینایی لبر (LHON) با جهش DNA میتوکندری و وراثت مادری. آتروفی بینایی با وراثت اتوزومال مغلوب شامل سندرم ولفرام است. هر دو ADOA و LHON دژنراسیون سلولهای گانگلیونی شبکیه (RGC) را به دلیل اختلال عملکرد میتوکندری به عنوان پاتولوژی مشترک دارند، اما تصویر بالینی و علل ژنتیکی متفاوت هستند6).
شیوع آن 1:12000 تا 1:50000 تخمین زده میشود و شایعترین نوروپاتی بینایی ارثی است2). شیوع کلی نوروپاتیهای بینایی ارثی در بریتانیا حدود 1:25000 گزارش شده است6).
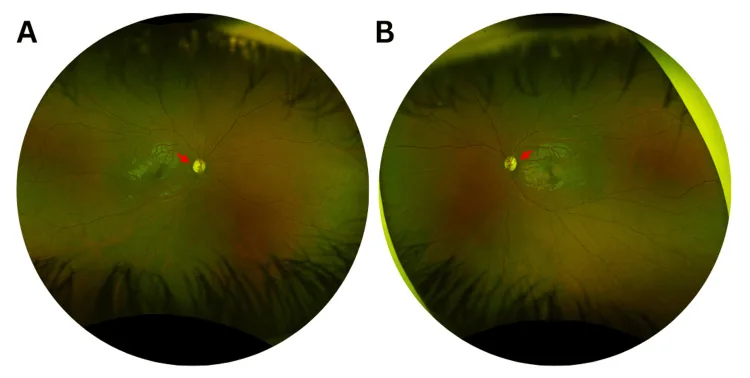
شروع معمول در دهه اول تا دوم زندگی است. به دلیل پیشرفت آهسته، بسیاری از بیماران نمیتوانند زمان دقیق شروع را مشخص کنند.
بله. در گزارش Tachibana و همکاران (2025)، یک جهش جدید در OPA1 در بیماری که در سن 56 سالگی بهترین حدت بینایی اصلاحشده 0.8/0.6 را حفظ کرده بود، شناسایی شد 2). نفوذ بین 43 تا 100٪ متغیر است و در ناقلان بدون علامت نیز ممکن است تنها با OCT ناهنجاری تشخیص داده شود.
سه یافته اصلی عبارتند از: سر عصب بینایی، بینایی رنگ و میدان بینایی.
ADOA علاوه بر آتروفی عصب بینایی، فنوتیپ DOA پلاس (DOA plus) با ناهنجاریهای سیستمیک دارد. ۲۰ تا ۳۰٪ بیماران دچار کاهش شنوایی، نوروپاتی محیطی، میوپاتی، آتاکسی، و افتالمپلژی خارجی مزمن پیشرونده (CPEO) میشوند3).
نوع کلاسیک (DOA)
علائم: کاهش تدریجی و دوطرفه بینایی.
یافتههای پاپیلاری: رنگپریدگی گوهای گیجگاهی یافته اصلی است.
علائم سیستمیک: فقط آتروفی عصب بینایی.
حدت بینایی: بیش از ۸۰٪ بیماران حدت بینایی ۰.۱ یا بهتر را حفظ میکنند.
DOA پلاس
فراوانی: در ۲۰ تا ۳۰٪ بیماران رخ میدهد3).
عوارض: کاهش شنوایی، نوروپاتی محیطی، میوپاتی، آتاکسی، CPEO و غیره.
نوع شدید: جهشهای دوآللی OPA1 باعث سندرم بهر (شروع زودرس، اختلال شدید بینایی، آتاکسی، تشنج) میشود3).
بیش از ۶۰٪ موارد ADOA ناشی از جهش در ژن OPA1 است6). OPA1 در موقعیت کروموزومی 3q28-q29 قرار دارد و بیش از ۵۰۰ جهش بیماریزا شناسایی شده است6). در ژاپن، جهش c.2708_2711 delTTAG به عنوان یک جهش شایع شناخته میشود.
در موارد منفی OPA1، باید به دنبال جهشهای ژنی دیگر گشت. ژنهای اصلی عامل در زیر آورده شدهاند.
| ژن | بیماری مرتبط | توضیحات |
|---|---|---|
| OPA1 | ADOA (نوع کلاسیک و DOA پلاس) | بیش از 60% موارد6) |
| AFG3L2 | آتروفی بینایی نوع 12 (OAT12)، SCA28 | حدود 3% از نوروپاتیهای ارثی بینایی1) |
| OPA3 | آتروفی بینایی غالب + آب مروارید و کمشنوایی (سندرم Costeff) | OMIM #258501 |
| WFS1 | شبه سندرم ولفرام | OMIM #222370, #614296 |
| DNM1L | آتروفی بینایی نوع 5 (OPA5) | کنترل تقسیم میتوکندری |
برادسکی و همکاران (2023) در یک پدر و دختر از تبار شرق آفریقا (سومالی) نوع 12 آتروفی بینایی ناشی از جهش c.1064C>T (p.Thr355Met) در ژن AFG3L2 را شناسایی کردند1). در یک مطالعه کوهورت روی 2186 مورد، AFG3L2 در میان 10 ژن برتر ایجادکننده نوروپاتی بینایی ارثی قرار داشت و 14 مورد از 451 مورد (3%) را شامل میشد.
بله. چندین ژن مانند AFG3L2، OPA3، WFS1 و DNM1L (OPA5) شناسایی شدهاند. به ویژه AFG3L2 حدود 3% از نوروپاتیهای بینایی ارثی را تشکیل میدهد1) و در صورت منفی بودن OPA1، جستجوی جامع با توالییابی اگزوم/ژنوم اهمیت دارد.
در صورت مشاهده اختلال پیشرفت بینایی دوطرفه با علت ناشناخته در سنین مدرسه، باید به این بیماری مشکوک شد. سابقه خانوادگی مشابه یک سرنخ مهم است، اما به دلیل نفوذ ناقص، ممکن است سابقه خانوادگی وجود نداشته باشد.
در مورد یک مرد 56 ساله گزارش شده توسط Tachibana و همکاران (2025)، پریمتری HFA 24-2 با محرک سفید طبیعی بود، اما پریمتری آبی-روی-زرد کاهش حساسیت را نشان داد2). OCT نازک شدن RNFL تمپورال را نشان داد و CFF به 30/31 هرتز (طبیعی >39 هرتز) کاهش یافت.
| بیماری | الگوی شروع | الگوی وراثت | نکات افتراقی |
|---|---|---|---|
| LHON | حاد تا تحت حاد | وراثت مادری | شایع در مردان جوان، شدید |
| ADOA | آهسته و پنهان | اتوزومال غالب | کودکی، دوطرفه و متقارن |
| گلوکوم | آهسته | چندعاملی | افزایش فشار داخل چشم، گود شدن پاپی |
| نوروپاتی فشاری بینایی | آهسته تا تحتحاد | غیرارثی | ضایعه کیاسمای بینایی در MRI |
تشخیصهای افتراقی دیگر: نوروپاتی سمی بینایی (اتامبوتول و غیره)، نوروپاتی تغذیهای بینایی، دیستروفی ماکولار پنهان، دیستروفی مخروطی، آمبلیوپی عملکردی، اختلال بینایی روانزاد.
هیچ درمان مؤثر ثابت شدهای وجود ندارد. مراقبت از کمبینایی و مشاوره با بیمار اساس درمان را تشکیل میدهد.
موارد زیر برای کاهش استرس اکسیداتیو پیشنهاد شدهاند، اما هیچکدام به عنوان درمان استاندارد تایید نشدهاند.
توضیح وراثت اتوزومال غالب و ارائه اطلاعات در مورد خطر شدت بیماری (سندرم بهر) ناشی از جهشهای دو آللی مهم است 3).
کاهش بینایی معمولاً به آرامی پیشرفت میکند و در مقایسه با LHON سیر خفیفتری دارد. بیش از 80٪ بیماران حدت بینایی اصلاحشده 0.1 یا بالاتر را حفظ میکنند، اما مواردی با کاهش به زیر 0.1 نیز وجود دارد. به دلیل شروع تدریجی، ممکن است دیر تشخیص داده شود و به طور اتفاقی در معاینات کشف شود. از آنجایی که درمان قطعی وجود ندارد، پایش منظم و مراقبت از کمبینایی برای مدیریت طولانیمدت مهم است.
در حال حاضر هیچ درمان مؤثر اثباتشدهای وجود ندارد. مراقبتهای کمبینایی اساس درمان را تشکیل میدهد. اگرچه گزارشهایی از احتمال تثبیت یا بهبود بینایی با ایبِدِنون وجود دارد4)5)، اما به عنوان درمان استاندارد تأیید نشده است. برای رویکردهای درمانی در مرحله تحقیق، به بخش «پژوهشهای جدید و چشمانداز آینده» مراجعه کنید.
OPA1 یک GTPase شبه دینامین است که در غشای داخلی میتوکندری قرار دارد. پس از سنتز در هسته، به میتوکندری منتقل میشود و وظایف زیر را بر عهده دارد6):
مکانیسم اصلی پاتولوژیک، هاپلوناکافی است. بیشتر جهشهای OPA1 باعث خاتمه زودرس ترجمه شده و منجر به کمبود پروتئین OPA1 میشوند6).
کاهش پروتئین OPA1 → افزایش قطعهقطعه شدن میتوکندری و افزایش بازیافت3) → اختلال متابولیسم میتوکندری و اختلال فسفریلاسیون اکسیداتیو → افزایش گونههای فعال اکسیژن (ROS) → آپوپتوز سلولهای گانگلیونی شبکیه (RGC).
عمدتاً سلولهای گانگلیونی شبکیه در باند پاپیلوماکولار دچار دژنراسیون میشوند که به صورت آتروفی عصب بینایی تمپورال ظاهر میشود.
AFG3L2 زیرواحد متالوپروتئاز AAA ماتریکس میتوکندری (m-AAA) را کد میکند1). با SPG7 (پاراپلژین) کمپلکس تشکیل داده و پردازش، بلوغ و کنترل کیفیت پروتئینهای میتوکندری را به صورت وابسته به ATP انجام میدهد. جهش در آن مشابه OPA1 باعث دژنراسیون سلولهای گانگلیونی شبکیه میشود1).
درمان ASO
هدف: اگزون القاکننده NMD (تجزیه وابسته به بیمعنی) در pre-mRNA OPA1.
مکانیسم: مهار درج اگزون القاکننده NMD و افزایش ترجمه OPA1 نوع وحشی از طریق یک رویکرد مستقل از جهش 6).
وضعیت فعلی: افزایش تولید پروتئین OPA1 و بهبود بیوانرژتیک میتوکندری در سه رده سلولی مشتق از بیماران ADOA تأیید شده است 6). نیاز به تزریق مکرر داخل زجاجیهای دارد و خطر اندوفتالمیت و یووئیت مزمن چالشهایی هستند.
ژن درمانی
مدل موشی: ژن درمانی OPA1 از دست رفتن سلولهای گانگلیونی شبکیه (RGC) را در مدل موشی DOA پیشگیری میکند 7).
بهینهسازی ایزوفرم: ایزوفرمهای بهینهشده OPA1 1 و 7 اثر درمانی را در مدلهای اختلال عملکرد میتوکندری نشان میدهند 8).
وضعیت فعلی: مرحله پیشبالینی.
ویرایش ژن
CRISPR-Cas9: اصلاح جهش OPA1 c.1334G>A: p.R445H در iPSC هموستاز میتوکندری را بازیابی میکند 9).
ترانساسپلایسینگ: رویکردهای اصلاح جهشهای بیماریزا در سطح mRNA نیز در دست بررسی هستند 6).
سلولهای بنیادی: بازسازی عصب بینایی با استفاده از سلولهای RGC مشتق از iPSC در مرحله تحقیقات پیشبالینی است6).
تجزیه و تحلیل مکرر توالییابی اگزوم نیز یک پیشرفت مهم تشخیصی است. با گسترش دانش ژنتیکی، ممکن است تشخیصهای جدیدی از دادههایی که در آزمایشهای قبلی منفی بودند، به دست آید1).