Аутосомно-доминантная атрофия зрительного нерва (ADOA) — это наследственная оптическая нейропатия, характеризующаяся двусторонней прогрессирующей атрофией зрительного нерва. Номер OMIM: 165500. Это самая распространенная наследственная оптическая нейропатия с предполагаемой распространенностью от 1:12 000 до 1:50 0002).
Наследственные оптические нейропатии делятся на две основные категории: ADOA, вызванная мутациями ядерных генов и менделевским наследованием, и наследственная оптическая нейропатия Лебера (LHON), вызванная мутациями митохондриальной ДНК и материнским наследованием. Аутосомно-рецессивная атрофия зрительного нерва включает синдром Вольфрама. ADOA и LHON имеют общий патогенез — дегенерацию ганглиозных клеток сетчатки (RGC) вследствие митохондриальной дисфункции, но различаются по клинической картине и генетическим причинам6).
QКак часто встречается аутосомно-доминантная атрофия зрительного нерва?
A
Распространенность оценивается от 1:12 000 до 1:50 000, что делает ее самой частой наследственной оптической нейропатией2). В Великобритании общая распространенность всех наследственных оптических нейропатий составляет около 1:25 0006).
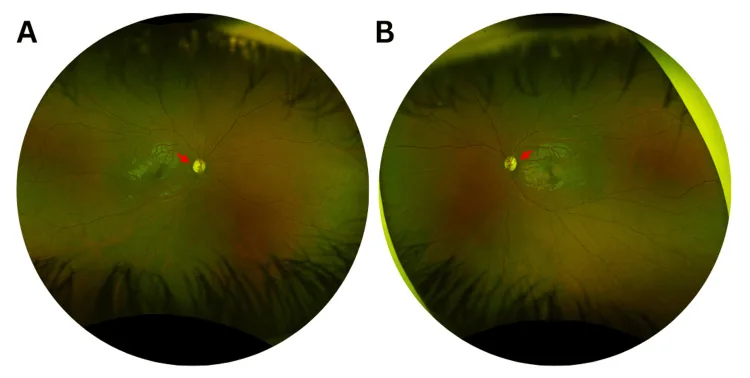
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
Цветные фотографии глазного дна правого (A) и левого (B) глаза при первом визите, демонстрирующие легкое височное побледнение диска зрительного нерва (красные стрелки) и расширение экскавации в обоих глазах. Это соответствует побледнению диска зрительного нерва, описанному в разделе «2. Основные симптомы и клинические признаки».
Типичное начало приходится на первое-второе десятилетие жизни. Из-за медленного прогрессирования многие пациенты не могут точно определить время начала.
Снижение остроты зрения: двустороннее, симметричное, с медленным и скрытым течением.
Повод для обнаружения: часто выявляется в школьном возрасте как нарушение развития бинокулярного зрения. Субъективные симптомы скудны, иногда обнаруживается случайно при профилактическом осмотре.
Степень остроты зрения: более 80% пациентов сохраняют остроту зрения не менее 20/200 (0,1), но в некоторых случаях корригированная острота зрения составляет 0,1 и ниже.
Пенетрантность: варьирует от 43 до 100% в зависимости от семьи2). Некоторые случаи сохраняют относительно хорошую зрительную функцию (максимальная корригированная острота зрения [BCVA] 0,6–1,0) до среднего возраста2).
Нет половых различий: возникновение не зависит от пола.
QВозможна ли ADOA при хорошем зрении?
A
Да. В отчете Tachibana и соавт. (2025) была идентифицирована новая мутация OPA1 у пациента, сохранявшего максимальную корригированную остроту зрения 0,8/0,6 в возрасте 56 лет2). Пенетрантность варьирует от 43 до 100%, и у бессимптомных носителей аномалии могут быть обнаружены только с помощью ОКТ.
Три основных признака: диск зрительного нерва, цветовое зрение и поле зрения.
Диск зрительного нерва: типична височная бледность клиновидной формы (temporal pallor). Также может наблюдаться диффузная атрофия.
Нарушение цветового зрения: может проявляться как приобретенная тританопия.
Дефекты поля зрения: центральная скотома, центроцекальная скотома и парацентральная скотома наиболее распространены. В некоторых случаях аномалии почти отсутствуют.
Сине-желтая периметрия: более чувствительна для выявления снижения чувствительности, чем стандартная автоматическая периметрия с белым стимулом2).
Оптическая когерентная томография (ОКТ): истончение внутренних слоев сетчатки, преимущественно папилломакулярного пучка. Характерно височное истончение слоя нервных волокон сетчатки (RNFL). Может наблюдаться микрокистозный макулярный отек (MME).
Электрофизиология: ЗВП показывают снижение амплитуды и удлинение латентности. Паттерн-электроретинограмма (pERG) показывает снижение компонента N95.
ADOA может иметь фенотип DOA plus (DOA plus) с системными нарушениями помимо атрофии зрительного нерва. У 20–30% пациентов наблюдаются сопутствующие нарушения слуха, периферическая нейропатия, миопатия, атаксия, хроническая прогрессирующая наружная офтальмоплегия (CPEO) и др. 3).
Типичная форма (DOA)
Симптомы: Двустороннее медленно прогрессирующее снижение зрения.
Изменения диска зрительного нерва: Височная клиновидная бледность является ключевым признаком.
Системные симптомы: Только атрофия зрительного нерва.
Острота зрения: Более 80% пациентов сохраняют остроту зрения ≥ 0,1.
DOA plus
Частота: Встречается у 20–30% пациентов 3).
Осложнения: Нарушения слуха, периферическая нейропатия, миопатия, атаксия, CPEO и др.
Тяжелая форма: Биаллельные мутации OPA1 приводят к синдрому Бера (раннее начало, тяжелое нарушение зрения, атаксия, судороги) 3).
Более 60% случаев ADOA вызваны мутациями гена OPA1 6). OPA1 расположен на хромосоме 3q28-q29, идентифицировано более 500 патогенных мутаций 6). В Японии мутация c.2708_2711 delTTAG известна как частая мутация.
Гаплонедостаточность: Большинство мутаций OPA1 вызывают преждевременную терминацию трансляции, что приводит к снижению количества белка OPA1 6).
Неполная пенетрантность: усложняет диагностику, прогнозирование и генетическое консультирование6).
De novo мутации: возникают с высокой частотой, поэтому отсутствие семейного анамнеза не исключает диагноз1).
Brodsky и соавт. (2023) идентифицировали атрофию зрительного нерва 12 типа, вызванную мутацией c.1064C>T (p.Thr355Met) гена AFG3L2, у отца и дочери восточноафриканского (сомалийского) происхождения1). В когортном исследовании 2186 случаев AFG3L2 входил в топ-10 генов наследственной оптической нейропатии, составляя 14 из 451 случая (3%).
QСуществуют ли другие гены-причины, кроме OPA1?
A
Да. Идентифицировано несколько генов, включая AFG3L2, OPA3, WFS1, DNM1L (OPA5). В частности, AFG3L2 составляет около 3% наследственных оптических нейропатий1), и при отрицательном результате по OPA1 важно провести исчерпывающий поиск с помощью экзомного/геномного секвенирования.
При выявленном в школьном возрасте двустороннем нарушении развития зрения неясной причины следует заподозрить данное заболевание. Семейный анамнез подобных симптомов является важной подсказкой, но из-за неполной пенетрантности семейный анамнез может отсутствовать.
Тест Фарнсворта-Манселла 100 оттенков: показывает ось тританопии.
Сине-желтая автоматическая периметрия: легче выявляет снижение чувствительности, чем стандартное белое поле зрения2).
ОКТ: оценивает истончение слоя нервных волокон сетчатки (RNFL) с преобладанием в височном и нижнем квадрантах. У бессимптомных носителей аномалии могут быть обнаружены только на ОКТ.
В случае 56-летнего мужчины, описанном Tachibana и соавт. (2025), белое поле зрения HFA 24-2 было нормальным, но сине-желтое поле зрения выявило снижение чувствительности2). ОКТ показала истончение височного RNFL, а КЧСМ была снижена до 30/31 Гц (норма >39 Гц).
Генетическое тестирование OPA1: необходимо для окончательного диагноза. Внешние тесты еще не широко распространены и требуют направления в специализированный центр.
Секвенирование экзома/генома: у OPA1-отрицательных пациентов важен поиск других генов, включая AFG3L2. Повторный анализ иногда может привести к новому диагнозу1).
Электрофизиологические тесты: ВП (снижение амплитуды, увеличение латентности), pERG (снижение N95), снижение КЧСМ.
ОКТА: полезна для оценки нейроваскулярных изменений макулы и перипапиллярной области.
Для снижения окислительного стресса были предложены следующие средства, но ни одно из них не является стандартной терапией.
Идебенон: синтетический аналог коэнзима Q10 (CoQ10). Он улучшает митохондриальное дыхание, минуя комплекс I электрон-транспортной цепи 6). Имеются сообщения о возможной стабилизации или улучшении остроты зрения у пациентов с ADOA с мутацией OPA1 4)5).
CoQ10, витамины B12, C, лютеин: предложены в качестве антиоксидантных добавок.
Снижение остроты зрения обычно прогрессирует медленно и протекает более мягко по сравнению с LHON. Более 80% пациентов сохраняют корригированную остроту зрения 0,1 и выше, но в некоторых случаях она может снижаться ниже 0,1. Из-за медленного начала заболевание может долго оставаться незамеченным или обнаруживаться случайно при профилактических осмотрах. Поскольку эффективного лечения не существует, важное значение имеют регулярное наблюдение и слабовидение для долгосрочного ведения.
QСуществует ли эффективное лечение ADOA?
A
В настоящее время не существует установленного эффективного лечения. Основой терапии является помощь при слабовидении (лоу-вижн кэр). Идебенон продемонстрировал потенциал для стабилизации или улучшения зрения в некоторых исследованиях4)5), но не утвержден в качестве стандартного лечения. Об экспериментальных подходах см. раздел «Последние исследования и перспективы».
OPA1 — это GTPаза, родственная динаміну, расположенная во внутренней мембране митохондрий. Она синтезируется в ядре, транспортируется в митохондрии и выполняет следующие функции6).
Слияние внутренней мембраны: поддержание митохондриальной сети
Поддержание структуры крист: стабилизация комплексов дыхательной цепи
Состав комплексов электрон-транспортной цепи: эффективность окислительного фосфорилирования
Основным патологическим механизмом является гаплонедостаточность. Большинство мутаций OPA1 приводят к преждевременной терминации трансляции, что снижает количество белка OPA16).
Снижение белка OPA1 → усиление фрагментации митохондрий и ускорение их рециклинга3) → нарушение метаболизма митохондрий и окислительного фосфорилирования → повышение уровня активных форм кислорода (АФК) → апоптоз ГКС.
В основном дегенерируют ГКС папилломакулярного пучка, что проявляется височной атрофией зрительного нерва.
AFG3L2 кодирует субъединицу митохондриальной матриксной AAA-металлопротеазы (m-AAA)1). Она образует комплекс с SPG7 (параплегином) и осуществляет процессинг, созревание и контроль качества митохондриальных белков АТФ-зависимым образом. Мутации вызывают дегенерацию ГКС, аналогичную OPA11).
7. Новейшие исследования и перспективы на будущее (отчёты исследовательской стадии)
Механизм : Ингибирование включения экзона, индуцирующего NMD, мутационно-независимый подход для усиления трансляции дикого типа OPA16).
Текущее состояние : Подтверждено увеличение продукции белка OPA1 и улучшение митохондриальной биоэнергетики в трёх клеточных линиях пациентов с ADOA6). Требуются регулярные интравитреальные инъекции, с рисками эндофтальмита и хронического увеита в качестве проблемы.
Генная терапия
Модель мыши : Генная терапия OPA1 предотвращает потерю RGC на мышиной модели DOA7).
Оптимизация изоформ : Оптимизированные изоформы OPA1 1 и 7 показывают терапевтический эффект на модели митохондриальной дисфункции8).
Транс-сплайсинг : Подход к коррекции патогенных мутаций на уровне мРНК также исследуется6).
Стволовые клетки: Регенерация зрительного нерва с помощью RGC, полученных из iPSC, изучается на доклиническом этапе6).
Повторный анализ данных экзомного секвенирования также является важным диагностическим достижением. С расширением генетических знаний из ранее отрицательных данных могут быть получены новые диагнозы1).
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.