विशिष्ट प्रकार (DOA)
लक्षण : द्विपक्षीय धीमी प्रगतिशील दृष्टि हानि।
पैपिला निष्कर्ष : टेम्पोरल वेज-आकार का पीलापन प्रमुख निष्कर्ष है।
प्रणालीगत लक्षण : केवल ऑप्टिक एट्रोफी।
दृष्टि : 80% से अधिक रोगी 0.1 या उससे अधिक दृष्टि बनाए रखते हैं।

ऑटोसोमल डोमिनेंट ऑप्टिक एट्रोफी (ADOA) एक वंशानुगत ऑप्टिक न्यूरोपैथी है जो द्विपक्षीय प्रगतिशील ऑप्टिक एट्रोफी द्वारा विशेषता है। OMIM संख्या 165500 है। यह सबसे आम वंशानुगत ऑप्टिक न्यूरोपैथी है, जिसका प्रसार 1:12,000 से 1:50,000 तक अनुमानित है2)।
वंशानुगत ऑप्टिक न्यूरोपैथी को मोटे तौर पर दो श्रेणियों में विभाजित किया गया है: ADOA, जो परमाणु जीन उत्परिवर्तन और मेंडेलियन वंशानुक्रम के कारण होता है, और लेबर वंशानुगत ऑप्टिक न्यूरोपैथी (LHON), जो माइटोकॉन्ड्रियल डीएनए उत्परिवर्तन और मातृ वंशानुक्रम के कारण होता है। ऑटोसोमल रिसेसिव ऑप्टिक एट्रोफी में वोल्फ्राम सिंड्रोम शामिल है। ADOA और LHON दोनों में माइटोकॉन्ड्रियल डिसफंक्शन के कारण रेटिनल गैंग्लियन कोशिका (RGC) अध:पतन का सामान्य रोग तंत्र है, लेकिन नैदानिक चित्र और आनुवंशिक कारण भिन्न हैं6)।
प्रसार 1:12,000 से 1:50,000 तक अनुमानित है, जो इसे सबसे आम वंशानुगत ऑप्टिक न्यूरोपैथी बनाता है2)। यूके में, सभी वंशानुगत ऑप्टिक न्यूरोपैथी का कुल प्रसार लगभग 1:25,000 बताया गया है6)।
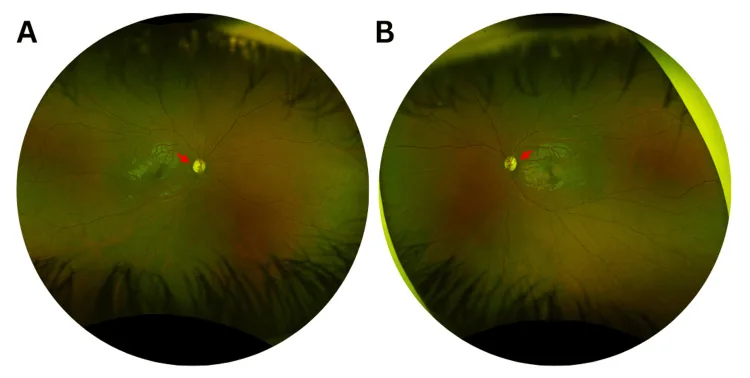
विशिष्ट शुरुआत जीवन के पहले से दूसरे दशक में होती है। धीमी प्रगति के कारण, कई रोगी सटीक शुरुआत का समय निर्धारित नहीं कर पाते।
हाँ। Tachibana एट अल. (2025) की रिपोर्ट में, 56 वर्ष की आयु में 0.8/0.6 की सर्वोत्तम सही दृष्टि बनाए रखने वाले एक मामले में OPA1 का नया उत्परिवर्तन पाया गया2)। प्रवेशन 43 से 100% तक भिन्न होता है, और लक्षणहीन वाहकों में केवल OCT द्वारा असामान्यताएं पाई जा सकती हैं।
तीन मुख्य निष्कर्ष हैं ऑप्टिक डिस्क, रंग दृष्टि और दृश्य क्षेत्र।
ADOA में ऑप्टिक एट्रोफी के अलावा अन्य प्रणालीगत असामान्यताओं के साथ DOA प्लस (DOA plus) फेनोटाइप होता है। 20-30% रोगियों में बहरापन, परिधीय न्यूरोपैथी, मायोपैथी, गतिभंग, क्रोनिक प्रोग्रेसिव एक्सटर्नल ऑप्थाल्मोप्लेजिया (CPEO) आदि सह-मौजूद होते हैं 3)।
विशिष्ट प्रकार (DOA)
लक्षण : द्विपक्षीय धीमी प्रगतिशील दृष्टि हानि।
पैपिला निष्कर्ष : टेम्पोरल वेज-आकार का पीलापन प्रमुख निष्कर्ष है।
प्रणालीगत लक्षण : केवल ऑप्टिक एट्रोफी।
दृष्टि : 80% से अधिक रोगी 0.1 या उससे अधिक दृष्टि बनाए रखते हैं।
DOA प्लस
आवृत्ति : 20-30% रोगियों में होता है 3)।
जटिलताएं : बहरापन, परिधीय न्यूरोपैथी, मायोपैथी, गतिभंग, CPEO आदि।
गंभीर प्रकार : बायएलिलिक OPA1 उत्परिवर्तन बेहर सिंड्रोम (प्रारंभिक शुरुआत, गंभीर दृष्टि हानि, गतिभंग, ऐंठन) का कारण बनता है 3)।
ADOA के 60% से अधिक मामले OPA1 जीन उत्परिवर्तन के कारण होते हैं 6)। OPA1 गुणसूत्र 3q28-q29 पर स्थित है और 500 से अधिक रोगजनक उत्परिवर्तन पहचाने गए हैं 6)। जापान में, c.2708_2711 delTTAG एक उच्च-आवृत्ति उत्परिवर्तन के रूप में जाना जाता है।
OPA1-नकारात्मक मामलों में अन्य जीन उत्परिवर्तनों की खोज आवश्यक है। मुख्य कारण जीन नीचे दिए गए हैं।
| जीन | संबंधित रोग | टिप्पणी |
|---|---|---|
| OPA1 | ADOA (विशिष्ट प्रकार, DOA प्लस) | कुल का 60% से अधिक6) |
| AFG3L2 | ऑप्टिक एट्रोफी प्रकार 12 (OAT12), SCA28 | वंशानुगत ऑप्टिक न्यूरोपैथी का लगभग 3%1) |
| OPA3 | प्रभावी ऑप्टिक एट्रोफी + मोतियाबिंद/बहरापन (कोस्टेफ सिंड्रोम) | OMIM #258501 |
| WFS1 | वोल्फ्राम सिंड्रोम जैसा | OMIM #222370, #614296 |
| DNM1L | ऑप्टिक एट्रोफी प्रकार 5 (OPA5) | माइटोकॉन्ड्रियल विभाजन नियंत्रण |
Brodsky एट अल. (2023) ने पूर्वी अफ्रीकी (सोमाली) मूल के पिता-पुत्री में AFG3L2 जीन c.1064C>T (p.Thr355Met) उत्परिवर्तन के कारण ऑप्टिक एट्रोफी प्रकार 12 की पहचान की1)। 2186 मामलों के कोहोर्ट अध्ययन में, वंशानुगत ऑप्टिक न्यूरोपैथी के शीर्ष 10 कारण जीनों में AFG3L2 शामिल था, जो 451 में से 14 मामलों (3%) में पाया गया।
हाँ। AFG3L2, OPA3, WFS1, DNM1L (OPA5) सहित कई जीनों की पहचान की गई है। विशेष रूप से, AFG3L2 वंशानुगत ऑप्टिक न्यूरोपैथी का लगभग 3% हिस्सा है1), और यदि OPA1 नकारात्मक है, तो एक्सोम/जीनोम अनुक्रमण द्वारा व्यापक खोज महत्वपूर्ण है।
स्कूली उम्र में पाई जाने वाली अज्ञात कारण की द्विपक्षीय दृष्टि विकास विकार में इस रोग का संदेह करें। समान लक्षणों का पारिवारिक इतिहास एक महत्वपूर्ण सुराग है, लेकिन अपूर्ण प्रवेश के कारण पारिवारिक इतिहास नहीं भी हो सकता है।
Tachibana एट अल. (2025) के 56 वर्षीय पुरुष मामले में, HFA 24-2 सफेद उत्तेजना दृश्य क्षेत्र सामान्य था, लेकिन नीले-पीले दृश्य क्षेत्र में संवेदनशीलता में कमी पाई गई2)। OCT ने टेम्पोरल RNFL पतलापन दिखाया, और CFF 30/31 Hz (सामान्य >39 Hz) तक कम हो गया।
| रोग | शुरुआत का तरीका | वंशानुक्रम पैटर्न | विभेदन बिंदु |
|---|---|---|---|
| LHON | तीव्र से अर्धतीव्र | मातृवंशीय | युवा पुरुषों में प्रमुख, गंभीर |
| ADOA | धीमा और गुप्त | ऑटोसोमल प्रभावी | बचपन, दोनों आँखों में सममित |
| ग्लूकोमा | धीमा | बहुक्रियात्मक | अंतर्नेत्र दबाव में वृद्धि, ऑप्टिक डिस्क कपिंग का बढ़ना |
| संपीडक ऑप्टिक न्यूरोपैथी | धीमा से अर्धतीव्र | अनुवांशिक नहीं | MRI में काइआज़्म घाव |
अन्य विभेदक निदान: विषाक्त ऑप्टिक न्यूरोपैथी (एथमब्यूटोल आदि), पोषण संबंधी ऑप्टिक न्यूरोपैथी, गुप्त मैक्यूलर डिस्ट्रोफी, कोन डिस्ट्रोफी, कार्यात्मक एम्ब्लियोपिया, मनोवैज्ञानिक दृश्य विकार।
कोई स्थापित प्रभावी उपचार नहीं है। कम दृष्टि देखभाल और रोगी परामर्श उपचार का मुख्य आधार है।
ऑक्सीडेटिव तनाव को कम करने के उद्देश्य से निम्नलिखित प्रस्तावित किए गए हैं, लेकिन इनमें से कोई भी मानक उपचार के रूप में स्थापित नहीं है।
ऑटोसोमल प्रभावी वंशानुक्रम की व्याख्या और द्विअलील उत्परिवर्तन के कारण गंभीरता (बेहर सिंड्रोम) के जोखिम के बारे में जानकारी प्रदान करना महत्वपूर्ण है 3)।
दृश्य तीक्ष्णता में कमी आमतौर पर धीरे-धीरे बढ़ती है और LHON की तुलना में हल्का कोर्स होता है। 80% से अधिक रोगी 0.1 या उससे अधिक की सही दृश्य तीक्ष्णता बनाए रखते हैं, लेकिन कुछ मामलों में यह 0.1 से नीचे गिर सकती है। धीमी शुरुआत के कारण जागरूकता में देरी हो सकती है और जांच में आकस्मिक खोज हो सकती है। स्थापित उपचार के अभाव में, दीर्घकालिक प्रबंधन के लिए नियमित निगरानी और लो विज़न केयर महत्वपूर्ण हैं।
वर्तमान में कोई स्थापित प्रभावी उपचार नहीं है। कम दृष्टि देखभाल (लो विज़न केयर) ही मुख्य उपचार है। आइडेबेनोन ने दृष्टि स्थिरीकरण या सुधार की संभावना दिखाई है4)5), लेकिन यह मानक उपचार के रूप में स्थापित नहीं है। शोध चरण के उपचार दृष्टिकोणों के लिए, “नवीनतम शोध और भविष्य की संभावनाएँ” अनुभाग देखें।
OPA1 एक डायनामिन-संबंधित GTPase है जो माइटोकॉन्ड्रिया की आंतरिक झिल्ली में स्थित होता है। यह केंद्रक में संश्लेषित होने के बाद माइटोकॉन्ड्रिया में परिवहनित होता है और निम्नलिखित कार्य करता है6)।
मुख्य रोग तंत्र हैप्लोअपर्याप्तता (haploinsufficiency) है। OPA1 उत्परिवर्तनों का अधिकांश भाग अनुवाद के समयपूर्व समाप्ति का कारण बनता है, जिससे OPA1 प्रोटीन की मात्रा कम हो जाती है6)।
OPA1 प्रोटीन में कमी → माइटोकॉन्ड्रियल विखंडन में वृद्धि और पुनर्चक्रण में तेजी3) → माइटोकॉन्ड्रियल चयापचय असामान्यता और ऑक्सीडेटिव फॉस्फोरिलीकरण में बाधा → प्रतिक्रियाशील ऑक्सीजन प्रजातियों (ROS) में वृद्धि → RGC का एपोप्टोसिस।
मुख्य रूप से पैपिलो-मैक्यूलर बंडल के RGC अध:पतित होते हैं, जो टेम्पोरल ऑप्टिक तंत्रिका शोष के रूप में प्रकट होता है।
AFG3L2 माइटोकॉन्ड्रियल मैट्रिक्स AAA मेटालोप्रोटीएज (m-AAA) की एक उपइकाई को कोड करता है1)। यह SPG7 (पैराप्लेजिन) के साथ एक जटिल बनाता है और ATP-निर्भर तरीके से माइटोकॉन्ड्रियल प्रोटीन के प्रसंस्करण, परिपक्वता और गुणवत्ता नियंत्रण करता है। उत्परिवर्तन OPA1 के समान RGC अध:पतन का कारण बनते हैं1)।
ASO थेरेपी
लक्ष्य : OPA1 pre-mRNA का NMD (नॉनसेंस-मध्यस्थ क्षरण) प्रेरक एक्सॉन।
तंत्र : NMD प्रेरित करने वाले एक्सॉन के समावेश को रोककर, जंगली प्रकार OPA1 अनुवाद को बढ़ाने वाला उत्परिवर्तन-स्वतंत्र दृष्टिकोण6)।
वर्तमान स्थिति : तीन ADOA रोगी कोशिका रेखाओं में OPA1 प्रोटीन उत्पादन में वृद्धि और माइटोकॉन्ड्रियल बायोएनर्जेटिक्स में सुधार की पुष्टि6)। नियमित रूप से कांचीय इंजेक्शन की आवश्यकता होती है, जिसमें एंडोफ्थैल्मिटिस और क्रोनिक यूवाइटिस का जोखिम चुनौतीपूर्ण है।
जीन थेरेपी
माउस मॉडल : OPA1 जीन थेरेपी DOA माउस मॉडल में RGC हानि को रोकती है7)।
आइसोफॉर्म अनुकूलन : अनुकूलित OPA1 आइसोफॉर्म 1 और 7 माइटोकॉन्ड्रियल डिसफंक्शन मॉडल में चिकित्सीय प्रभाव दिखाते हैं8)।
वर्तमान स्थिति : प्रीक्लिनिकल चरण।
जीन संपादन
CRISPR-Cas9 : iPSC में OPA1 c.1334G>A: p.R445H उत्परिवर्तन का सुधार माइटोकॉन्ड्रियल होमियोस्टेसिस को बहाल करता है9)।
ट्रांस-स्प्लिसिंग : mRNA स्तर पर रोगजनक उत्परिवर्तन सुधार का दृष्टिकोण भी शोधाधीन है6)।
स्टेम कोशिकाएं : iPSC-व्युत्पन्न RGC द्वारा ऑप्टिक तंत्रिका पुनर्जनन का प्रीक्लिनिकल अध्ययन चल रहा है6)।
एक्सोम अनुक्रमण का बार-बार पुनर्विश्लेषण भी निदान में एक महत्वपूर्ण प्रगति है। आनुवंशिक ज्ञान के विस्तार के साथ, पिछले परीक्षणों में नकारात्मक डेटा से नए निदान प्राप्त हो सकते हैं1)।