A Atrofia Óptica Dominante Autossômica (Autosomal Dominant Optic Atrophy; ADOA) é uma neuropatia óptica hereditária caracterizada por atrofia óptica progressiva bilateral. O número OMIM é 165500. É a neuropatia óptica hereditária mais frequente, com prevalência estimada entre 1:12.000 e 1:50.000 2).
As neuropatias ópticas hereditárias são amplamente divididas em ADOA (genes nucleares, herança mendeliana) e Neuropatia Óptica Hereditária de Leber (LHON) (mutações no DNA mitocondrial, herança materna). A atrofia óptica recessiva autossômica inclui a síndrome de Wolfram. Tanto ADOA quanto LHON compartilham a mesma patologia de degeneração das células ganglionares da retina (RGC) devido à disfunção mitocondrial, mas os quadros clínicos e as causas genéticas são diferentes 6).
QQual a frequência da Atrofia Óptica Dominante Autossômica?
A
A prevalência é estimada entre 1:12.000 e 1:50.000, sendo a mais frequente entre as neuropatias ópticas hereditárias 2). A prevalência geral de neuropatias ópticas hereditárias no Reino Unido foi relatada como cerca de 1:25.000 6).
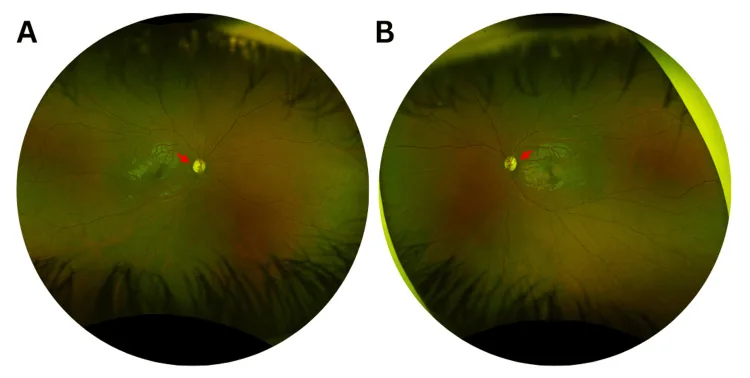
Murati Calderon RA, et al. Clinical and Genetic Findings in an Autosomal Dominant Optic Atrophy-Compatible Phenotype Harboring an OPA1 Variant: A Case Report. Cureus. 2025. Figure 1. PMCID: PMC12659938. License: CC BY.
Fotografias coloridas do fundo de olho direito (A) e esquerdo (B) na primeira consulta, mostrando palidez temporal leve do disco óptico (setas vermelhas) e alargamento da escavação do disco óptico em ambos os olhos. Isso corresponde à palidez do disco óptico discutida na seção “2. Principais Sintomas e Achados Clínicos”.
O início típico ocorre na primeira ou segunda década de vida. Devido à progressão lenta, muitos pacientes não conseguem determinar o momento exato do início.
Diminuição da visão: Curso bilateral, simétrico, lento e insidioso.
Momento da descoberta: Frequentemente descoberto na idade escolar como um distúrbio do desenvolvimento da visão binocular. Os sintomas subjetivos são escassos e pode ser descoberto incidentalmente em exames de rotina.
Nível de visão: Mais de 80% dos pacientes mantêm visão de 20/200 (0,1) ou melhor, mas alguns casos apresentam acuidade visual corrigida de 0,1 ou menos.
Penetrância: Varia de 43 a 100% dependendo da família 2). Alguns casos mantêm função visual relativamente boa (melhor acuidade visual corrigida de 0,6 a 1,0) até a meia-idade 2).
Sem diferença de gênero: Não há diferença na ocorrência entre homens e mulheres.
QÉ possível ter ADOA mesmo com boa visão?
A
Sim. No relato de Tachibana et al. (2025), uma nova mutação no OPA1 foi identificada em um caso que manteve a melhor acuidade visual corrigida de 0,8/0,6 aos 56 anos 2). A penetrância varia de 43 a 100%, e anormalidades podem ser detectadas apenas na OCT em portadores assintomáticos.
Os três achados principais são disco óptico, visão de cores e campo visual.
Disco óptico: Palidez em forma de cunha (palidez temporal) é típica, com atrofia difusa também observada.
Anomalia da visão de cores: Pode apresentar tritanopia adquirida.
Defeito de campo visual: Escotoma central, escotoma papilocentral e escotoma paracentral são os mais comuns. Alguns casos podem não apresentar quase nenhuma anormalidade.
Campo visual azul-amarelo: Detecta diminuição da sensibilidade mais facilmente do que a campimetria automatizada com estímulo branco comum 2).
Tomografia de Coerência Óptica (OCT): Afinamento das camadas internas da retina, principalmente no feixe papilomacular. O afinamento temporal da camada de fibras nervosas da retina (CFNR) é característico. Pode ser observado edema macular microcístico (EMM).
Eletrofisiologia: O VEP mostra amplitude reduzida e latência prolongada. No eletrorretinograma de padrão (pERG), observa-se redução do componente N95.
A ADOA apresenta um fenótipo DOA plus com anormalidades sistêmicas além da atrofia do nervo óptico. 20-30% dos pacientes apresentam complicações como perda auditiva, neuropatia periférica, miopatia, ataxia e oftalmoplegia externa progressiva crônica (CPEO) 3).
Mais de 60% dos casos de ADOA são causados por mutações no gene OPA1 6). OPA1 está localizado no cromossomo 3q28-q29, e mais de 500 mutações patogênicas foram identificadas 6). No Japão, a mutação c.2708_2711 delTTAG é conhecida como uma mutação de alta frequência.
Haploinsuficiência: A maioria das mutações do OPA1 causa terminação precoce da tradução, resultando em quantidade insuficiente da proteína OPA1 6).
Penetrância incompleta: Complexifica o diagnóstico, a predição prognóstica e o aconselhamento genético6).
Mutação de novo: Ocorre com alta frequência, portanto o diagnóstico não pode ser excluído mesmo sem história familiar 1).
Em casos negativos para OPA1, é necessário pesquisar outras mutações genéticas. Abaixo estão os principais genes causadores.
Gene
Doença associada
Observações
OPA1
ADOA (tipo típico / DOA plus)
Mais de 60% do total 6)
AFG3L2
Atrofia óptica tipo 12 (OAT12), SCA28
Cerca de 3% das neuropatias ópticas hereditárias 1)
OPA3
Atrofia óptica dominante com catarata / perda auditiva (síndrome de Costeff)
OMIM #258501
WFS1
Semelhante à síndrome de Wolfram
OMIM #222370, #614296
DNM1L
Atrofia óptica tipo 5 (OPA5)
Controle da divisão mitocondrial
Brodsky et al. (2023) identificaram atrofia óptica tipo 12 devido à mutação c.1064C>T (p.Thr355Met) no gene AFG3L2 em um pai e filha de ascendência do Leste Africano (Somali)1). Em um estudo de coorte de 2186 casos, AFG3L2 estava entre os 10 principais genes causadores de neuropatia óptica hereditária, representando 14 de 451 casos (3%).
QExistem outros genes causadores além do OPA1?
A
Sim. Vários genes foram identificados, como AFG3L2, OPA3, WFS1 e DNM1L (OPA5). Particularmente, AFG3L2 representa cerca de 3% dos casos de neuropatia óptica hereditária1), e quando OPA1 é negativo, a busca abrangente por sequenciamento de exoma/genoma é importante.
Se for encontrada deficiência visual bilateral inexplicada em idade escolar, suspeite desta doença. Histórico familiar de sintomas semelhantes é uma pista importante, mas pode não haver histórico familiar devido à penetrância incompleta.
Teste Farnsworth-Munsell 100 hue: Mostra um eixo de tritanopia.
Campimetria automatizada blue-on-yellow: Detecta redução de sensibilidade mais facilmente que a campimetria normal com estímulo branco2).
OCT: Avalia afinamento da RNFL predominante nos quadrantes temporal e inferior. Mesmo em portadores assintomáticos, anormalidades podem ser detectadas apenas com OCT.
No caso de um homem de 56 anos de Tachibana et al. (2025), a campimetria HFA 24-2 com estímulo branco era normal, mas a campimetria blue-on-yellow detectou redução de sensibilidade2). A OCT mostrou afinamento temporal da RNFL, e a CFF reduziu para 30/31 Hz (normal >39 Hz).
Teste genético OPA1: Necessário para diagnóstico definitivo. Testes externos ainda não são difundidos, sendo necessário encaminhamento para centros de referência.
Sequenciamento de exoma/genoma: Em casos negativos para OPA1, a busca por outros genes incluindo AFG3L2 é importante. A reanálise pode fornecer um novo diagnóstico1).
Para reduzir o estresse oxidativo, os seguintes foram propostos, mas nenhum estabelecido como terapia padrão.
Idebenona: Análogo sintético da coenzima Q10 (CoQ10). Melhora a respiração mitocondrial ao desviar do complexo I da cadeia de transporte de elétrons 6). Há relatos de possível estabilização ou melhora da visão em pacientes com ADOA e mutação OPA1 4)5).
CoQ10, vitamina B12, C, luteína: Propostos como suplementos antioxidantes.
É importante explicar a herança autossômica dominante e fornecer informações sobre o risco de gravidade devido a mutações bialélicas (síndrome de Behr) 3).
A perda visual geralmente progride lentamente e é mais branda em comparação com LHON. Mais de 80% dos pacientes mantêm acuidade visual corrigida de 0,1 ou melhor, mas há casos que caem para 0,1 ou menos. Como o início é lento, a percepção pode ser tardia, sendo às vezes descoberto acidentalmente em exames. Como não há terapia estabelecida, o monitoramento regular e os cuidados de longo prazo com baixa visão são importantes.
QExiste terapia eficaz para ADOA?
A
Atualmente, não há terapia eficaz estabelecida. O cuidado com baixa visão é o principal tratamento. A idebenona mostrou potencial para estabilizar ou recuperar a visão em alguns relatos 4)5), mas não é um tratamento padrão estabelecido. Para abordagens terapêuticas em fase de pesquisa, consulte a seção “Pesquisas Recentes e Perspectivas Futuras”.
OPA1 é uma GTPase relacionada à dinamina localizada na membrana interna mitocondrial. Sintetizada no núcleo e transportada para a mitocôndria, desempenha as seguintes funções 6).
Fusão da membrana interna: Manutenção da rede mitocondrial
Manutenção da estrutura das cristas: Estabilização dos complexos da cadeia respiratória
Formação de complexos de transporte de elétrons: Eficiência da fosforilação oxidativa
O principal mecanismo patológico é a haploinsuficiência. A maioria das mutações no OPA1 causa terminação precoce da tradução, resultando em quantidade insuficiente da proteína OPA1 6).
Redução da proteína OPA1 → Aumento da fragmentação mitocondrial e aumento da reciclagem 3) → Disfunção metabólica mitocondrial e prejuízo da fosforilação oxidativa → Aumento de espécies reativas de oxigênio (ROS) → Apoptose das células ganglionares da retina (RGC).
Principalmente as células ganglionares da retina no feixe papilomacular degeneram, manifestando-se como atrofia do nervo óptico temporal.
AFG3L2 codifica a subunidade da metaloprotease AAA da matriz mitocondrial (m-AAA) 1). Forma um complexo com SPG7 (paraplegina) e realiza o processamento, maturação e controle de qualidade de proteínas mitocondriais de forma dependente de ATP. Mutações causam degeneração das RGCs semelhante ao OPA1 1).
7. Pesquisas Recentes e Perspectivas Futuras (Relatórios em Fase de Pesquisa)
Alvo: Éxon indutor de NMD (degradação dependente de nonsense) do pré-mRNA de OPA1.
Mecanismo: Inibe a inclusão do éxon que induz NMD, aumentando a tradução de OPA1 do tipo selvagem por uma abordagem independente de mutação 6).
Estado Atual: Aumento da produção da proteína OPA1 e melhora da bioenergética mitocondrial confirmados em três linhagens celulares derivadas de pacientes com ADOA 6). Requer injeções intravítreas repetidas regularmente, com riscos de endoftalmite e uveíte crônica como desafios.
Terapia Gênica
Modelo Murino: Terapia gênica com OPA1 previne a perda de células ganglionares da retina em modelo murino de DOA 7).
Otimização de Isoformas: As isoformas otimizadas 1 e 7 de OPA1 mostram efeito terapêutico em modelos de disfunção mitocondrial 8).
Estado Atual: Fase pré-clínica.
Edição Gênica
CRISPR-Cas9: A correção da mutação OPA1 c.1334G>A: p.R445H em iPSC restaura a homeostase mitocondrial 9).
Trans-splicing: Abordagens de correção de mutações patogênicas em nível de mRNA também estão sendo estudadas 6).
Células-tronco: A regeneração do nervo óptico usando RGC derivadas de iPSC está sendo investigada em estudos pré-clínicos 6).
A reanálise repetida do sequenciamento do exoma também é um avanço diagnóstico importante. Com a expansão do conhecimento genético, novos diagnósticos podem ser obtidos a partir de dados que eram negativos em exames anteriores 1).
Brodsky MC, Olson RJ, Asumda FZ, Lopour MQR, Schimmenti LA, Klee EW. Identification of AFG3L2 dominant optic atrophy following reanalysis of clinical exome sequencing. American journal of ophthalmology case reports. 2023;30:101825. doi:10.1016/j.ajoc.2023.101825. PMID:36974169; PMCID:PMC10038781.
Tachibana M, Hayashi T, Igawa Y, Mizobuchi K, Miyasaka Y, Tsuchihashi T, et al. Case of autosomal dominant optic atrophy with relatively good visual function. BMC ophthalmology. 2025;25(1):443. doi:10.1186/s12886-025-04276-5. PMID:40751186; PMCID:PMC12315315.
Othman BA, Ong JE, Dumitrescu AV. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes. 2022;13(6). doi:10.3390/genes13061005. PMID:35741767; PMCID:PMC9223020.
Barboni P, Valentino ML, La Morgia C, Carbonelli M, Savini G, De Negri A, et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain : a journal of neurology. 2013;136(Pt 2):e231. doi:10.1093/brain/aws280. PMID:23388408.
Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Annals of clinical and translational neurology. 2020;7(4):590-594. doi:10.1002/acn3.51026. PMID:32243103; PMCID:PMC7187718.
Wong DCS, Makam R, Yu-Wai-Man P. Advanced therapies for inherited optic neuropathies. Eye (London, England). 2026;40(2):177-184. doi:10.1038/s41433-025-04109-1. PMID:41318849; PMCID:PMC12830909.
Sarzi E, Seveno M, Piro-Mégy C, et al. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci Rep. 2018;8:2468. doi:10.1038/s41598-018-20838-8.
Maloney DM, Chadderton N, Millington-Ward S, Palfi A, Shortall C, O’Byrne JJ, et al. Optimized OPA1 Isoforms 1 and 7 Provide Therapeutic Benefit in Models of Mitochondrial Dysfunction. Frontiers in neuroscience. 2020;14:571479. doi:10.3389/fnins.2020.571479. PMID:33324145; PMCID:PMC7726421.
Sladen PE, Perdigão PRL, Salsbury G, Novoselova T, van der Spuy J, Chapple JP, et al. CRISPR-Cas9 correction of OPA1 c.1334G>A: p.R445H restores mitochondrial homeostasis in dominant optic atrophy patient-derived iPSCs. Molecular therapy. Nucleic acids. 2021;26:432-443. doi:10.1016/j.omtn.2021.08.015. PMID:34589289; PMCID:PMC8455316.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.