La consulenza genetica è un servizio medico il cui obiettivo è «fornire informazioni genetiche corrette». Fornisce a pazienti e famiglie informazioni sulla diagnosi, sul pattern di ereditarietà, sul rischio di ricorrenza, sugli esami disponibili e sulle opzioni di trattamento delle malattie genetiche, e sostiene decisioni autonome. Questa definizione è condivisa anche a livello internazionale e i tre pilastri della consulenza genetica sono «fornitura di informazioni», «supporto psicologico» e «supporto decisionale»3).
Le malattie oculari ereditarie rappresentano circa il 43% della disabilità visiva congenita1). La frequenza delle anomalie cromosomiche nei figli è di circa 0,5-1%. La discromatopsia rosso-verde è una delle malattie oculari ereditarie più comuni, presente in circa il 5% degli uomini e nello 0,2% delle donne. La prevalenza della retinite pigmentosa è di circa 1/4.000-1/5.000, ed è una delle principali cause di disabilità visiva2).
La consulenza genetica non riguarda solo il paziente con la malattia ereditaria, ma anche i familiari che potrebbero զարգ? No. We’ll use: che potrebbero svilupparla e i portatori preoccupati di trasmetterla ai futuri figli. L’oftalmologo, mentre si occupa della diagnosi, collabora con specialisti in genetica e consulenti genetici certificati per fornire informazioni.
QDove si può ricevere la consulenza genetica?
A
Può essere ricevuta nei reparti di medicina genetica degli ospedali universitari o degli ospedali di riferimento, oppure in strutture con consulenti genetici certificati. Le strutture possono essere trovate consultando gli elenchi dei consulenti genetici certificati della Società Giapponese di Consulenza Genetica e della Società Giapponese di Genetica Umana. Anche il sito del Centro informazioni sulle malattie rare fornisce indicazioni su dove consultare.
2. Modalità di ereditarietà e principali malattie oculari ereditarie
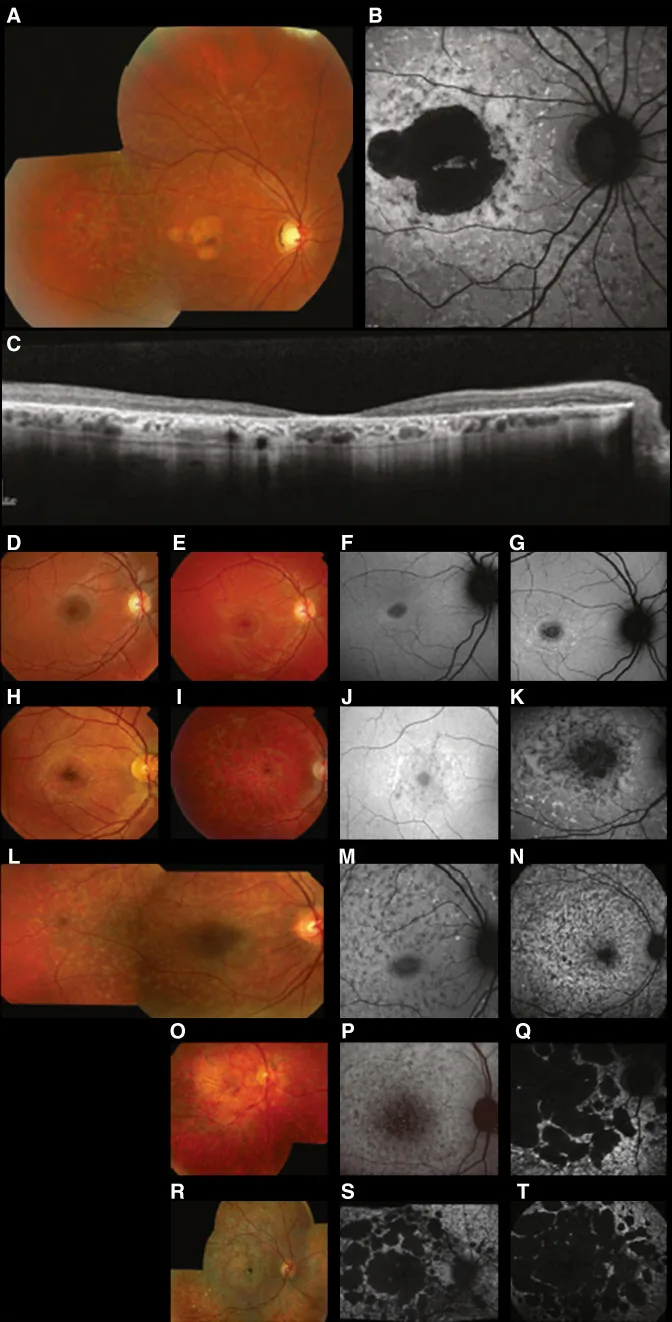
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Immagini multimodali tipiche della malattia di Stargardt (STGD1): la fotografia a colori del fundus mostra macchie giallo-bianche a livello dell’epitelio pigmentato retinico e atrofia maculare (A); l’autofluorescenza del fundus mostra un’area maculare ipoautofluorescente con fluorescenza anomala intorno (B); e la SD-OCT mostra una marcata perdita degli strati retinici esterni e dell’EPR, con focolai iperriflettenti corrispondenti alle macchie (C). Corrisponde alla malattia di Stargardt (una distrofia retinica autosomica recessiva causata da mutazioni del gene ABCA4) trattata nella sezione “2. Modalità di ereditarietà e principali malattie oculari ereditarie”.
Il modello di ereditarietà è un’informazione centrale nella consulenza genetica e si classifica in quattro modelli principali.
Ereditarietà autosomica dominante
Condizione di insorgenza: La malattia compare con una mutazione in un allele (stato eterozigote).
Caratteristiche dell’albero genealogico: I membri affetti compaiono in generazioni consecutive.
Rischio di recidiva: La probabilità di trasmetterla a un figlio è del 50%.
Nota: Se la penetranza non è del 100%, può verificarsi il salto di una generazione.
Ereditarietà autosomica recessiva
Condizione di insorgenza: La malattia compare quando entrambi gli alleli sono mutati (omozigote o eterozigote composto).
Caratteristiche dell’albero genealogico: I membri affetti compaiono tra fratelli e sorelle. I genitori sono di solito portatori (eterozigoti).
Rischio di recidiva: La probabilità di malattia in un figlio di due portatori è del 25%.
Tendenza recente: Poiché i matrimoni tra cugini sono diventati meno frequenti, la proporzione di eterozigoti composti è aumentata.
Ereditarietà legata all’X
Condizione di comparsa: La maggior parte dei pazienti è maschile (emizigote).
Nelle donne: Poiché hanno due cromosomi X, una mutazione in uno di essi le rende portatrici.
Caratteristiche dell’albero genealogico: I casi sono concentrati nei maschi e la trasmissione avviene dalla madre al figlio.
Rischio di ricorrenza: La probabilità che un figlio maschio di una madre portatrice sia colpito è del 50%.
Ereditarietà materna (ereditarietà mitocondriale)
Caratteristica: Il DNA mitocondriale (mtDNA) dello spermatozoo viene quasi interamente degradato al momento della fecondazione. Per questo si trasmette solo dalla madre al figlio.
QSe un genitore ha una malattia oculare ereditaria, qual è la probabilità che venga trasmessa al figlio?
A
Il tipo di ereditarietà varia. Nell’ereditarietà autosomica dominante, la probabilità di trasmissione da un genitore affetto al figlio è del 50%. Nell’ereditarietà autosomica recessiva, se entrambi i genitori sono portatori, la probabilità che un figlio sviluppi la malattia è del 25%. Nell’ereditarietà legata al cromosoma X, la probabilità che un figlio maschio di una madre portatrice sia affetto è del 50%, mentre nell’ereditarietà materna (mitocondriale), se la madre è affetta o portatrice, può essere trasmessa a tutti i figli. Per situazioni individuali, si raccomanda di consultare uno specialista in genetica o un consulente genetico certificato.
3. Cause e fattori di rischio delle malattie oculari ereditarie
I meccanismi di insorgenza delle malattie oculari ereditarie sono classificati in base al tipo di effetto che una mutazione genetica ha sulla funzione delle proteine.
Aploinsufficienza (haploinsufficiency): Il fenotipo compare quando si perde la funzione di un solo allele. Un esempio tipico è l’aniridia congenita (mutazione di PAX6). Un solo allele normale non riesce a fornire la quantità di prodotto genico necessaria per la normale formazione dei tessuti.
Effetto dominante negativo (dominant negative effect): La proteina mutata inibisce la funzione della proteina normale in modo competitivo e strutturale. Nella sindrome di Marfan (mutazione di FBN1), le molecole mutate di fibrillina-1 ostacolano la formazione della matrice extracellulare.
Mutazioni mitocondriali: Nella neuropatia ottica ereditaria di Leber (LHON), tre mutazioni puntiformi — 11778, 3460 e 14484 — rappresentano circa il 90% di tutte le mutazioni. Il difetto nella produzione di energia danneggia le cellule gangliari della retina.
Mutazioni de novo: Mutazioni non presenti nei genitori e che insorgono ex novo durante la formazione dell’ovocita o dello spermatozoo. Un esempio rappresentativo è l’amaurosi congenita di Leber causata da mutazioni di CRX. Possono comparire anche come casi isolati quando nessun altro nell’albero genealogico è affetto.
Eterozigote composto: Nelle malattie autosomiche recessive, è una forma in cui si verificano due mutazioni diverse nei due alleli. Con il recente calo dei matrimoni tra cugini, la proporzione di eterozigoti composti è aumentata rispetto agli omozigoti.
Disomia uniparentale: fenomeno in cui entrambe le copie dello stesso cromosoma sono ereditate da un solo genitore, mentre il cromosoma dell’altro genitore manca. Può presentarsi come un caso apparentemente sporadico10).
Sono stati identificati più di 100 geni causali per la retinite pigmentosa, e lo stesso fenotipo può essere causato anche da molti geni diversi2). Questa diversità genetica rende difficile la diagnosi.
4. Aspetti pratici della consulenza genetica e del test genetico
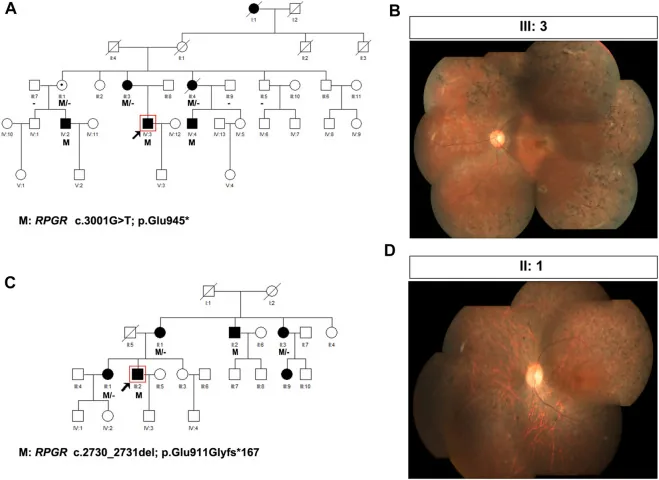
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Pedigree e fotografie del fundus di due famiglie con retinite pigmentosa legata all’X (varianti di RPGR: c.3001G>T e c.2730_2731del). I pedigree usano simboli standard (quadrati pieni per i maschi affetti, cerchi tratteggiati per le femmine portatrici) per mostrare il modello di trasmissione, e le fotografie del fundus corrispondenti mostrano una pigmentazione a spicole ossee avanzata e atrofia retinica. Questo corrisponde alla costruzione del pedigree e alla determinazione del modello di trasmissione trattate nella sezione “4. La pratica della consulenza genetica e dei test genetici”.
Per svolgere in modo appropriato la consulenza genetica, è necessaria la seguente preparazione.
Creazione di un pedigree: raccogliere l’anamnesi familiare di almeno tre generazioni e registrarla come pedigree. La distribuzione verticale e orizzontale dei soggetti affetti consente di stimare il modello di trasmissione.
Stima del modello di trasmissione: in base al pedigree e al fenotipo, inferire se si tratta di trasmissione autosomica dominante, autosomica recessiva, legata all’X o materna.
Spiegazione del test genetico e ottenimento del consenso scritto: prima di esaminare le informazioni genetiche del paziente, è necessario spiegare in modo adeguato il significato e i limiti del test e ottenere il consenso scritto.
Anonimizzazione dei risultati del test del DNA e gestione delle informazioni: proteggere la privacy mediante un’anonimizzazione collegabile.
Definizione preventiva della gestione dei reperti incidentali: confermare con il paziente, prima del test, la politica di comunicazione nel caso vengano rilevati reperti incidentali gravi potenzialmente letali.
Alta precisione. Adatto a confermare siti di mutazione noti
Test panel (sequenziamento mirato)
Gruppo di geni associati a malattie specifiche
Ricerca simultanea dei geni مرتبطی con le malattie della retina. Alto tasso diagnostico5)
Analisi dell’esoma (NGS)
Tutte le regioni esoniche
Rileva varianti sconosciute. Utile quando ci sono molti geni causa di malattia
Analisi dell’intero genoma (WGS)
Intero genoma
Può avere un tasso diagnostico più alto rispetto ai test NGS mirati esistenti13)
Nella distrofia retinica ereditaria, quando si sospetta una malattia causata da una mutazione del gene RPE65, e in casi come l’aiuto nel determinare l’idoneità alla terapia genica, alcuni esami che soddisfano i requisiti possono essere eseguiti come prestazioni sanitarie coperte dall’assicurazione4).
Interpretazione delle varianti e principali database
L’interpretazione delle varianti richiede un giudizio attento. È noto che circa il 30% delle descrizioni pubblicate come “varianti” negli articoli è in realtà costituito da polimorfismi (varianti normali). In generale, le variazioni della sequenza che compaiono in 1 o più persone ogni 100 soggetti sani dovrebbero essere considerate polimorfismi.
Si utilizzano i seguenti principali database.
OMIM (Online Mendelian Inheritance in Man): database completo delle malattie e dei geni ereditari
GeneReviews: fornisce informazioni di consulenza genetica per ciascuna malattia
RetNet (Retinal Information Network): database genetico specializzato nelle malattie della retina
QIl costo del test genetico è coperto dall’assicurazione?
A
In alcune malattie oculari ereditarie, il test genetico può essere eseguito come prestazione sanitaria coperta dall’assicurazione quando viene stabilita l’indicazione al trattamento e sono soddisfatti i requisiti della struttura designata. Tuttavia, la copertura varia in base al tipo di esame e alla malattia, quindi è necessario verificare presso la struttura in cui ci si reca. Gli esami non coperti dall’assicurazione possono essere a carico del paziente. Per le malattie designate come rare e difficili da trattare (come la retinite pigmentosa), può essere disponibile anche un sostegno economico tramite il sistema di sussidio per le spese mediche per le malattie designate come rare e difficili da trattare4).
5. Sistema di consulenza genetica e prospettive terapeutiche
Nell’erogazione della consulenza genetica, la collaborazione con uno specialista in genetica e un consulente genetico certificato è considerata ideale. Negli ospedali universitari può essere richiesta la valutazione del comitato ეთico. Poiché le informazioni genetiche possono influenzare non solo il paziente ma anche i familiari, nella loro gestione è richiesta particolare attenzione. Sta inoltre avanzando il dibattito sociale sulla prevenzione della discriminazione ingiusta basata sulle informazioni genetiche8).
Malattie rare designate e sostegno alle spese mediche
Per i pazienti con malattie rare, la quota a carico delle spese mediche viene ridotta. È fissato un tetto mensile per il totale di visite ambulatoriali, ricoveri e farmaci, e si applicano fasce in base al reddito.
Il farmaco di terapia genica voretigene neparvovec per la distrofia retinica ereditaria causata da mutazioni del gene RPE65 è una formulazione per somministrazione sottoretinica che utilizza un vettore AAV2. Efficacia e sicurezza sono state confermate in studi randomizzati controllati6). La decisione sull’indicazione del trattamento richiede la conferma del tipo di malattia tramite test genetico.
Per la terapia con oligonucleotidi antisenso (ASO), la somministrazione intravitreale di una formulazione per l’amaurosi congenita di Leber di tipo 10 (LCA10) causata da mutazioni di CEP290 è oggetto di studio in trial clinici9).
Il trapianto autologo di cellule dell’epitelio pigmentato retinico con cellule iPS è studiato come medicina rigenerativa per la degenerazione maculare legata all’età7). Pur essendo un ambito diverso dalle malattie retiniche ereditarie, attira attenzione come esempio concreto di terapia cellulare della retina.
QLa terapia genica è disponibile ora?
A
Nella distrofia retinica ereditaria causata da mutazione di RPE65, efficacia e sicurezza di voretigene neparvovec sono state dimostrate negli RCT6). Per confermare l’indicazione al trattamento, è necessario definire il tipo di malattia con un test genetico. Nella LCA10 con mutazione di CEP290, l’iniezione intravitreale di preparati ASO è in studio in trial clinici9).
L’ereditarietà autosomica dominante, autosomica recessiva e legata all’X segue le leggi di Mendel. Tuttavia, i fattori seguenti possono rendere difficile una previsione semplice.
Penetranza: anche se una persona porta la mutazione, non tutti sviluppano la malattia. Quando la penetranza è bassa, il disturbo può saltare generazioni, rendendo difficile dedurre il modello di ereditarietà dall’albero genealogico.
Espressività: anche tra familiari con lo stesso gene mutato, la gravità dei sintomi può variare.
Mutazione gain-of-function: meccanismo in cui la proteina mutata acquisisce una nuova funzione dannosa. È diverso dal solito effetto dominante negativo.
I mitocondri si trovano nel citoplasma e solo il mtDNA proveniente dall’ovocita materno viene trasmesso al figlio. In ogni cellula sono presenti migliaia di copie di mtDNA e può verificarsi una condizione in cui mtDNA mutato e mtDNA normale coesistono (eteroplasmia). Più alta è la proporzione di eteroplasmia, più gravi tendono a essere i sintomi. Nella neuropatia ottica ereditaria di Leber (LHON), tre varianti—11778 (la più comune), 3460 e 14484—rappresentano circa il 90% di tutte le varianti.
La disomia uniparentale è una condizione in cui entrambi i cromosomi di una coppia provengono dallo stesso genitore e manca il cromosoma dell’altro genitore. Un figlio nato da un portatore di una malattia autosomica recessiva può sviluppare la malattia anche se l’altro genitore non è portatore, dando l’apparenza di un caso sporadico10). Insieme alle varianti de novo e agli eterozigoti composti, una malattia ereditaria va presa in considerazione anche in assenza di anamnesi familiare.
Nella LCA10 con varianti di CEP290, la correzione dello splicing mediante terapia ASO è oggetto di studio in trial clinici9).
Il candidato alla terapia di editing genomico EDIT-101, basato su CRISPR/Cas9, è stato segnalato in fase di sviluppo preclinico per la LCA10 (varianti di CEP290)11).
Il test genetico preimpianto (PGT-M) può essere eseguito per malattie ereditarie autosomiche dominanti e recessive e può essere considerato all’interno di un quadro etico12).
È stato dimostrato che il sequenziamento dell’intero genoma (WGS) può aumentare il tasso di diagnosi molecolare nelle malattie retiniche ereditarie rispetto ai test genetici standard esistenti13).
Sono in fase di sviluppo strumenti di previsione della patogenicità delle varianti genetiche basati sull’intelligenza artificiale (AI), e si prevede un miglioramento dell’accuratezza nell’interpretazione delle varianti.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.