Генетическое консультирование — это медицинская услуга, цель которой состоит в том, чтобы «предоставлять правильную генетическую информацию». Пациентам и их семьям сообщают о диагнозе, типе наследования, риске повторного рождения, доступных обследованиях и методах лечения генетических заболеваний, а также помогают принимать самостоятельные решения. Это определение признано и на международном уровне; тремя опорами генетического консультирования являются «предоставление информации», «психологическая поддержка» и «поддержка в принятии решений»3).
Наследственные заболевания глаз составляют около 43% врожденных нарушений зрения1). Частота хромосомных аномалий у потомства составляет примерно 0,5–1%. Красно-зеленая цветовая слепота — одно из самых распространенных наследственных заболеваний глаз; она встречается примерно у 5% мужчин и 0,2% женщин. Распространенность пигментного ретинита составляет примерно 1/4 000–1/5 000, и это одна из основных причин нарушения зрения2).
К генетическому консультированию относятся не только сами пациенты с наследственным заболеванием, но и члены семьи, у которых оно может развиться, а также носители, которые беспокоятся о передаче его будущим детям. Офтальмолог, занимаясь диагностикой, также взаимодействует со специалистами по генетике и сертифицированными генетическими консультантами для предоставления информации.
QГде можно пройти генетическое консультирование?
A
Его можно пройти в генетических отделениях университетских больниц или крупных больниц, а также в учреждениях, где работают сертифицированные генетические консультанты. Учреждения можно найти по спискам сертифицированных генетических консультантов Японского общества генетического консультирования и Японского общества человеческой генетики. На сайте Информационного центра по редким заболеваниям также есть сведения о местах консультаций.
2. Типы наследования и основные наследственные заболевания глаз
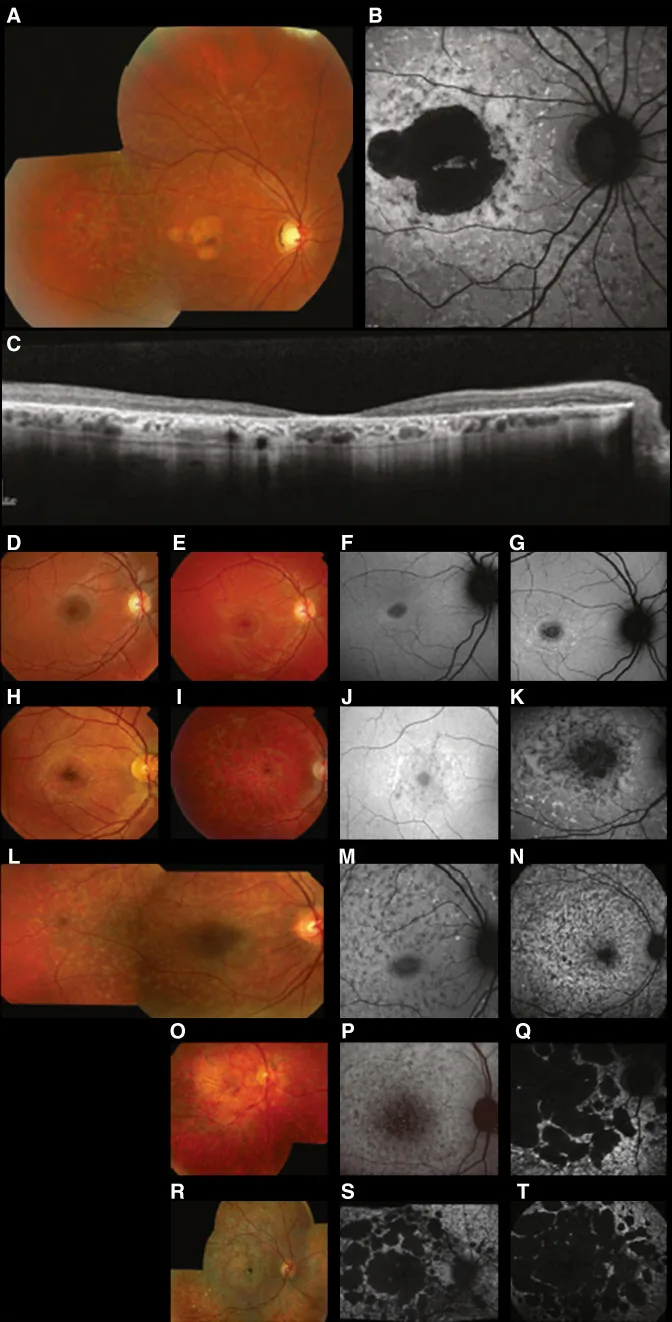
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Типичные мультимодальные изображения при болезни Старгардта (STGD1): цветная фотография глазного дна показывает желтовато-белые пятна на уровне пигментного эпителия сетчатки и атрофию макулы (A); аутофлуоресценция глазного дна показывает участок макулы с пониженной флуоресценцией и аномальную флуоресценцию вокруг него (B); а SD-OCT показывает выраженную утрату наружных слоев сетчатки и РПЭ, а также гиперрефлективные очаги, соответствующие пятнам (C). Это соответствует болезни Старгардта (аутосомно-рецессивной дистрофии сетчатки, вызванной мутациями гена ABCA4), о которой говорится в разделе «2. Типы наследования и основные наследственные заболевания глаз».
Тип наследования — это ключевая информация в генетическом консультировании, и его делят на четыре основных типа.
Аутосомно-доминантное наследование
Условие появления: Заболевание возникает при мутации в одном аллеле (состояние гетерозиготы).
Особенности родословной: Больные появляются в последовательных поколениях.
Риск повторного рождения: Вероятность передачи ребенку составляет 50%.
Примечание: Если пенетрантность не 100%, может быть пропуск поколения.
Аутосомно-рецессивное наследование
Условие появления: Заболевание возникает, когда оба аллеля мутированы (гомозигота или сложная гетерозигота).
Особенности родословной: Больные появляются среди братьев и сестер. Родители обычно являются носителями (гетерозиготами).
Риск повторного рождения: Вероятность заболевания у ребенка от двух носителей составляет 25%.
Недавняя тенденция: По мере уменьшения числа браков между двоюродными родственниками доля сложных гетерозигот увеличилась.
Сцепленное с X наследование
Условие проявления: Большинство пациентов — мужчины (гемизиготы).
У женщин: Поскольку у них две X-хромосомы, мутация в одной из них делает их носительницами.
Особенности родословной: Среди заболевших преобладают мужчины, и передача идет от матери к сыну.
Риск повторного возникновения: Вероятность того, что сын матери-носительницы будет поражен, составляет 50%.
Особенность: Митохондриальная ДНК (mtDNA) сперматозоида при оплодотворении почти полностью разрушается. Поэтому она передается только от матери к ребенку.
Пример заболевания: Наследственная оптическая нейропатия Лебера (LHON).
Отличие от сцепленного с X наследования: Различие в том, что встречаются и женщины-пациентки.
Гетероплазмия: Когда нормальная mtDNA и мутантная mtDNA сосуществуют, фенотип может различаться.
зернистая, решетчатая и авеллино дистрофия роговицы
аутосомно-доминантное
TGFBI (5q)
хрусталик
врожденная катаракта
аутосомно-доминантное (несколько генов)
—
хрусталик
синдром Марфана
аутосомно-доминантный
FBN1 (15q)
сетчатка
пигментный ретинит
аутосомно-доминантный, аутосомно-рецессивный и сцепленный с X-хромосомой
более 100 причинных генов2)
сетчатка
болезнь Штаргардта
аутосомно-рецессивный
ABCA4 (1p)
сетчатка
ювенильный ретиношизис
Х-сцепленный
RS1 (Xp)
сетчатка
ретинобластома
аутосомно-доминантный
RB1 (13q)
зрительный нерв
наследственная оптическая нейропатия Лебера
материнское наследование (mtDNA)
—
зрительный нерв
аутосомно-доминантная атрофия зрительного нерва
аутосомно-доминантный
OPA1 (3q)
цветовое зрение
красно-зелёное нарушение цветоощущения
сцепленный с Х-хромосомой
—
QЕсли у одного из родителей есть наследственное заболевание глаз, какова вероятность передать его ребенку?
A
Тип наследования различается. При аутосомно-доминантном наследовании вероятность передачи от больного родителя ребенку составляет 50%. При аутосомно-рецессивном наследовании, если оба родителя являются носителями, вероятность заболевания у ребенка составляет 25%. При Х-сцепленном наследовании вероятность заболевания у сына носительницы составляет 50%, а при материнском наследовании (митохондриальном), если мать больна или является носителем, заболевание может передаться всем детям. В конкретных ситуациях рекомендуется обратиться к врачу-генетику или сертифицированному генетическому консультанту.
3. Причины и факторы риска наследственных заболеваний глаз
Механизмы возникновения наследственных заболеваний глаз классифицируются по типу влияния, которое генетическая мутация оказывает на функцию белка.
Гаплонедостаточность (haploinsufficiency): Фенотип возникает уже при утрате функции только одного аллеля. Классический пример — врождённая аниридия (мутация PAX6). Одного нормального аллеля недостаточно, чтобы обеспечить нужное количество продукта гена для нормального формирования тканей.
Доминантно-негативный эффект (dominant negative effect): Мутантный белок конкурентно и структурно подавляет функцию нормального белка. При синдроме Марфана (мутация FBN1) мутантные молекулы фибриллина-1 нарушают формирование внеклеточного матрикса.
Митохондриальные мутации: При наследственной оптической нейропатии Лебера (LHON) три точечные мутации — 11778, 3460 и 14484 — составляют около 90% всех мутаций. Нарушение выработки энергии повреждает ганглиозные клетки сетчатки.
Мутации de novo: Мутации, которых нет у родителей и которые возникают заново при образовании яйцеклетки или сперматозоида. Типичный пример — врождённый амавроз Лебера, связанный с мутациями CRX. Они также могут встречаться как единичные случаи, когда больше никто в родословной не болен.
Составной гетерозигот: При аутосомно-рецессивных заболеваниях это вариант, при котором на двух аллелях возникают разные мутации. На фоне недавнего снижения числа браков между двоюродными родственниками доля составных гетерозигот увеличилась по сравнению с гомозиготами.
Однородительская дисомия: явление, при котором обе копии одной и той же хромосомы наследуются от одного родителя, а хромосома другого родителя отсутствует. Может проявляться как, казалось бы, спорадический случай10).
Для пигментного ретинита выявлено более 100 причинных генов, и даже один и тот же фенотип может быть обусловлен многими разными генами2). Такое генетическое разнообразие затрудняет диагностику.
4. Практические аспекты генетического консультирования и генетического тестирования
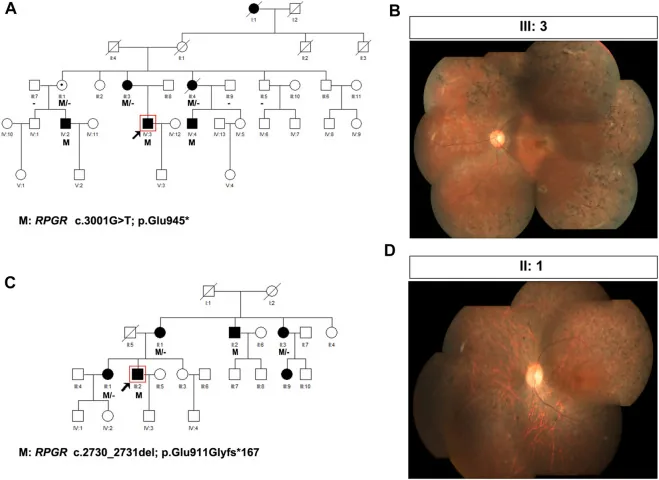
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Родословные и фотографии глазного дна двух семей с Х-сцепленной пигментной дегенерацией сетчатки (варианты RPGR: c.3001G>T и c.2730_2731del). В родословных используются стандартные символы (закрашенные квадраты для поражённых мужчин, пунктирные круги для женщин-носительниц), чтобы показать тип наследования, а соответствующие фотографии глазного дна демонстрируют выраженную пигментацию по типу костных остатков и атрофию сетчатки. Это соответствует составлению родословной (pedigree) и определению типа наследования, рассматриваемым в разделе «4. Практика генетического консультирования и генетического тестирования».
Для надлежащего проведения генетического консультирования необходима следующая подготовка.
Составление родословной (pedigree): Собрать семейный анамнез как минимум за три поколения и зафиксировать его в виде родословной. Вертикальное и горизонтальное распределение больных позволяет предположить тип наследования.
Определение типа наследования: По родословной и фенотипу определить, является ли наследование аутосомно-доминантным, аутосомно-рецессивным, Х-сцепленным или материнским.
Объяснение генетического теста и получение письменного согласия: Перед изучением генетической информации пациента необходимо подробно объяснить значение и ограничения теста и получить письменное согласие.
Анонимизация результатов ДНК-теста и управление информацией: Защищать конфиденциальность с помощью анонимизации с возможностью связи.
Заранее определить порядок действий при случайных находках: До тестирования согласовать с пациентом порядок сообщения, если будут выявлены серьёзные случайные находки, способные угрожать жизни.
Высокая точность. Подходит для подтверждения известных участков мутаций
Панельное тестирование (таргетное секвенирование)
Группа генов, связанных с определенными заболеваниями
Одновременный поиск генов, связанных с заболеваниями сетчатки. Высокая диагностическая эффективность5)
Анализ экзома (NGS)
Все экзонные области
Выявляет неизвестные варианты. Полезно, когда есть много генов, вызывающих заболевание
Анализ всего генома (WGS)
Весь геном
Может иметь более высокую диагностическую эффективность, чем существующие целевые NGS-тесты13)
При наследственной дистрофии сетчатки, когда подозревают заболевание, вызванное мутацией гена RPE65, а также в случаях, например, для помощи в определении показаний к генной терапии, некоторые исследования, соответствующие условиям, могут проводиться как медицинская помощь, покрываемая страховкой4).
Интерпретация вариантов требует осторожной оценки. Известно, что примерно 30% описаний, опубликованных в статьях как «варианты», на самом деле являются полиморфизмами (нормальными вариантами). В общем случае изменения последовательности нуклеотидов, встречающиеся у 1 и более из каждых 100 здоровых людей, следует рассматривать как полиморфизмы.
Используются следующие основные базы данных.
OMIM (Online Mendelian Inheritance in Man): комплексная база данных по наследственным заболеваниям и генам
GeneReviews: предоставляет информацию по генетическому консультированию для каждого заболевания
RetNet (Retinal Information Network): база данных генов, специализированная на заболеваниях сетчатки
QПокрываются ли расходы на генетическое тестирование страховкой?
A
При некоторых наследственных заболеваниях глаз генетическое тестирование может проводиться как медицинская помощь, покрываемая страховкой, если определены показания к лечению и выполнены требования назначенного учреждения. Однако покрытие зависит от типа исследования и заболевания, поэтому необходимо уточнять в учреждении, куда вы обращаетесь. Исследования, не покрываемые страховкой, могут выполняться за собственный счёт. При заболеваниях, отнесённых к редким и трудноизлечимым (например, пигментный ретинит), может также быть доступна финансовая поддержка по системе субсидирования медицинских расходов для таких заболеваний4).
5. Система генетического консультирования и перспективы лечения
При проведении генетического консультирования идеальным считается сотрудничество со специалистом по генетике и сертифицированным генетическим консультантом. В университетских больницах может потребоваться рассмотрение этическим комитетом. Поскольку генетическая информация может влиять не только на самого пациента, но и на членов семьи, при обращении с ней требуется особая осторожность. Также продолжается общественное обсуждение мер по предотвращению несправедливой дискриминации на основе генетической информации8).
Назначенные редкие заболевания и помощь с медицинскими расходами
Следующие наследственные заболевания глаз отнесены к редким заболеваниям и подлежат помощи с медицинскими расходами.
Пигментный ретинит
Врожденная глаукома
Желатинозная каплеобразная дистрофия роговицы
У пациентов с редкими заболеваниями снижается личная доля оплаты медицинских расходов. Установлен ежемесячный лимит на сумму амбулаторной помощи, госпитализации и отпуска лекарств, и применяются категории в зависимости от дохода.
Препарат генной терапии voretigene neparvovec для наследственной дистрофии сетчатки, вызванной мутациями гена RPE65, представляет собой средство для субретинального введения с использованием вектора AAV2. Его эффективность и безопасность подтверждены в рандомизированных контролируемых исследованиях6). Для решения вопроса о показаниях к лечению необходимо подтвердить тип заболевания с помощью генетического тестирования.
Что касается терапии антисмысловыми олигонуклеотидами (ASO), в клинических испытаниях изучается внутриглазное введение препарата для болезни Лебера, врожденной слепоты 10 типа (LCA10), вызванной мутациями CEP2909).
Аутологичная трансплантация клеток пигментного эпителия сетчатки с использованием iPS-клеток изучается как регенеративная медицина для возрастной макулярной дегенерации7). Хотя это иная область, чем наследственные заболевания сетчатки, она привлекает внимание как реальный пример клеточной терапии сетчатки.
QДоступна ли сейчас генная терапия?
A
При наследственной дистрофии сетчатки, вызванной мутацией RPE65, эффективность и безопасность voretigene neparvovec были показаны в РКИ6). Чтобы подтвердить, подходит ли лечение, нужно определить тип заболевания с помощью генетического теста. При LCA10 с мутацией CEP290 интравитреальное введение препаратов ASO изучается в клинических исследованиях9).
Аутосомно-доминантное, аутосомно-рецессивное и Х-сцепленное наследование подчиняются законам Менделя. Однако следующие факторы могут затруднять простое прогнозирование.
Пенетрантность: даже если человек несет мутацию, заболевают не все. При низкой пенетрантности признак может пропускать поколения, и по родословной трудно определить тип наследования.
Экспрессивность: даже у членов семьи с одним и тем же мутантным геном тяжесть симптомов может различаться.
Мутация с приобретением функции: механизм, при котором мутантный белок приобретает новую вредную функцию. Это отличается от обычного доминантно-отрицательного эффекта.
Митохондрии находятся в цитоплазме, и ребенку передается только mtDNA, полученная из яйцеклетки матери. В каждой клетке есть тысячи копий mtDNA, и может возникать состояние, когда мутантная mtDNA и нормальная mtDNA сосуществуют (гетероплазмия). Чем выше доля гетероплазмии, тем тяжелее, как правило, симптомы. При наследственной оптической нейропатии Лебера (LHON) три варианта — 11778 (наиболее частый), 3460 и 14484 — составляют около 90% всех вариантов.
Однородительская дисомия — это состояние, при котором обе хромосомы пары получены от одного и того же родителя, а хромосомы другого родителя отсутствуют. У ребенка, рожденного от носителя аутосомно-рецессивного заболевания, болезнь может развиться даже если другой родитель не является носителем, и это может выглядеть как спорадический случай10). Вместе с de novo-мутациями и составными гетерозиготами наследственное заболевание следует учитывать даже при отсутствии семейного анамнеза.
При LCA10 с вариантами CEP290 коррекция сплайсинга с помощью терапии ASO изучается в клинических испытаниях9).
Кандидат генной терапии EDIT-101 с использованием CRISPR/Cas9 был описан в доклинической разработке для LCA10 (варианты CEP290)11).
Предимплантационное генетическое тестирование (PGT-M) может проводиться при аутосомно-доминантных и аутосомно-рецессивных наследственных заболеваниях и может рассматриваться в рамках этических норм12).
Показано, что полногеномное секвенирование (WGS) может повышать молекулярную диагностическую частоту при наследственных заболеваниях сетчатки по сравнению с существующими стандартными генетическими тестами13).
Разрабатываются инструменты на основе искусственного интеллекта (AI) для прогнозирования патогенности генетических вариантов, и ожидается повышение точности интерпретации вариантов.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.