Genetische Beratung ist eine medizinische Dienstleistung mit dem Ziel, die richtigen genetischen Informationen zu vermitteln. Sie informiert Patienten und Familien über Diagnose, Erbgang, Wiederholungsrisiko, verfügbare Untersuchungen und Behandlungsmöglichkeiten genetischer Erkrankungen und unterstützt eine selbstbestimmte Entscheidungsfindung. Diese Definition ist auch international anerkannt; die drei Säulen der genetischen Beratung sind «Informationsvermittlung», «psychosoziale Unterstützung» und «Entscheidungsunterstützung»3).
Erbliche Augenerkrankungen machen etwa 43 % der angeborenen Sehbehinderungen aus1). Die Häufigkeit von Chromosomenanomalien bei Nachkommen liegt bei etwa 0,5 bis 1 %. Die Rot-Grün-Sehschwäche ist eine der häufigsten erblichen Augenerkrankungen und tritt bei etwa 5 % der Männer und 0,2 % der Frauen auf. Die Prävalenz der Retinitis pigmentosa liegt bei etwa 1/4.000 bis 1/5.000 und ist eine der Hauptursachen für Sehbehinderung2).
Zur genetischen Beratung gehören nicht nur die betroffenen Patienten selbst, sondern auch Familienmitglieder, bei denen die Erkrankung auftreten könnte, sowie Träger, die befürchten, sie an ihre zukünftigen Kinder weiterzugeben. Augenärzte übernehmen die Diagnose und arbeiten dabei mit Humangenetikern und zertifizierten genetischen Beratern zusammen, um Informationen zu vermitteln.
QWo kann man genetische Beratung erhalten?
A
Sie kann in den genetischen Abteilungen von Universitätskliniken oder Schwerpunktkrankenhäusern oder in Einrichtungen mit zertifizierten genetischen Beratern in Anspruch genommen werden. Einrichtungen lassen sich über die Verzeichnisse zertifizierter genetischer Berater der Japanischen Gesellschaft für Genetische Beratung und der Japanischen Gesellschaft für Humangenetik finden. Auch die Website des Nanbyo-Informationszentrums bietet Hinweise auf Beratungsstellen.
2. Vererbungsmuster und wichtige erbliche Augenerkrankungen
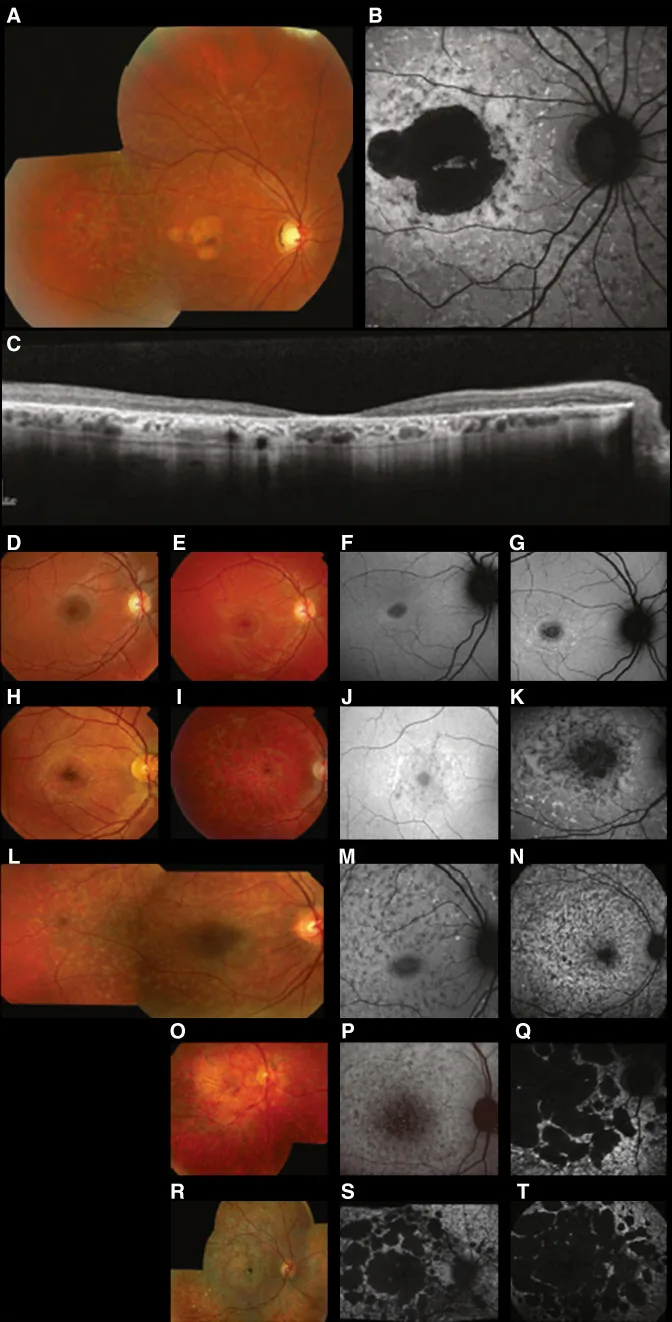
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Typische multimodale Bildgebung beim Stargardt-Syndrom (STGD1): Das Farbfundusfoto zeigt gelblich-weiße Flecken auf Höhe des retinalen Pigmentepithels und eine Makulaatrophie (A); die Fundusautofluoreszenz zeigt eine hypoautofluoreszente Makulazone mit abnormer Fluoreszenz darum herum (B); und die SD-OCT zeigt einen deutlichen Verlust der äußeren Netzhautschichten und des RPE mit hyperreflektiven Herden, die den Flecken entsprechen (C). Dies entspricht dem Stargardt-Syndrom (einer autosomal-rezessiven retinalen Dystrophie durch ABCA4-Mutationen), das im Abschnitt „2. Vererbungsmuster und wichtige erbliche Augenerkrankungen“ behandelt wird.
Das Vererbungsmuster ist eine Kerninformation in der genetischen Beratung und wird in vier Hauptmuster eingeteilt.
Autosomal-dominanter Erbgang
Auftretensbedingung: Die Erkrankung tritt bei einer Mutation in einem Allel auf (heterozygoter Zustand).
Merkmale des Stammbaums: Betroffene treten in aufeinanderfolgenden Generationen auf.
Wiederholungsrisiko: Die Wahrscheinlichkeit, es an ein Kind weiterzugeben, beträgt 50%.
Hinweis: Wenn die Penetranz nicht 100% beträgt, kann eine Generation übersprungen werden.
Autosomal-rezessiver Erbgang
Auftretensbedingung: Die Erkrankung tritt auf, wenn beide Allele mutiert sind (homozygot oder compound-heterozygot).
Merkmale des Stammbaums: Betroffene treten unter Geschwistern auf. Die Eltern sind meist Träger (heterozygot).
Wiederholungsrisiko: Die Wahrscheinlichkeit für eine Erkrankung bei einem Kind von zwei Trägern beträgt 25%.
Aktueller Trend: Da Heiraten zwischen Cousins seltener geworden sind, hat der Anteil compound-heterozygoter Personen zugenommen.
X-chromosomale Vererbung
Manifestationsbedingung: Die meisten Patienten sind männlich (hemizygot).
Bei Frauen: Da sie zwei X-Chromosomen haben, führt eine Mutation auf einem davon dazu, dass sie Trägerinnen sind.
Merkmale des Stammbaums: Betroffene sind überwiegend männlich, und die Weitergabe erfolgt von der Mutter an den Sohn.
Wiederholungsrisiko: Die Wahrscheinlichkeit, dass der Sohn einer Trägerin betroffen ist, beträgt 50 %.
Mütterliche Vererbung (mitochondriale Vererbung)
Merkmal: Die mitochondriale DNA (mtDNA) der Spermien wird bei der Befruchtung fast vollständig abgebaut. Daher wird sie nur von der Mutter an das Kind weitergegeben.
QWenn ein Elternteil eine erbliche Augenerkrankung hat, wie hoch ist die Wahrscheinlichkeit, dass sie an das Kind weitergegeben wird?
A
Das Vererbungsmuster ist unterschiedlich. Bei autosomal-dominanter Vererbung beträgt die Wahrscheinlichkeit, dass ein betroffenes Elternteil die Erkrankung an das Kind weitergibt, 50 %. Bei autosomal-rezessiver Vererbung liegt die Wahrscheinlichkeit, dass ein Kind erkrankt, bei 25 %, wenn beide Eltern Träger sind. Bei X-chromosomaler Vererbung beträgt die Wahrscheinlichkeit, dass ein Sohn einer Trägerin betroffen ist, 50 %, und bei mütterlicher Vererbung (mitochondrial) kann sie weitergegeben werden, wenn die Mutter betroffen ist oder Trägerin ist, an alle Kinder. Für den Einzelfall wird die Beratung durch eine Fachärztin oder einen Facharzt für Genetik oder eine zertifizierte genetische Beratung empfohlen.
3. Ursachen und Risikofaktoren erblich bedingter Augenerkrankungen
Die Entstehungsmechanismen erblich bedingter Augenerkrankungen werden nach der Art der Auswirkungen einer genetischen Veränderung auf die Proteinfunktion eingeteilt.
Haploinsuffizienz (haploinsufficiency): Der Phänotyp tritt auf, wenn die Funktion nur eines Allels verloren geht. Ein typisches Beispiel ist die kongenitale Aniridie (PAX6-Mutation). Ein normales Allel allein kann nicht genug Genprodukt für eine normale Gewebebildung bereitstellen.
Dominant-negativer Effekt (dominant negative effect): Das mutierte Protein hemmt die Funktion des normalen Proteins kompetitiv und strukturell. Beim Marfan-Syndrom (FBN1-Mutation) stören mutierte Fibrillin-1-Moleküle die Bildung der extrazellulären Matrix.
Mitochondriale Mutationen: Bei der Leber hereditären Optikusneuropathie (LHON) machen drei Punktmutationen — 11778, 3460 und 14484 — etwa 90 % aller Mutationen aus. Eine gestörte Energieproduktion schädigt die retinalen Ganglienzellen.
De-novo-Mutationen: Mutationen, die bei den Eltern nicht vorhanden sind und neu bei der Bildung von Ei- oder Samenzellen entstehen. Ein typisches Beispiel ist die Leber’sche kongenitale Amaurose durch CRX-Mutationen. Sie können auch als Einzelfälle auftreten, wenn sonst niemand in der Familienanamnese betroffen ist.
Compound-Heterozygot: Bei autosomal-rezessiven Erkrankungen ist dies eine Form, bei der zwei verschiedene Mutationen auf den beiden Allelen auftreten. Mit dem jüngsten Rückgang von Verwandtenehen ist der Anteil von Compound-Heterozygoten im Vergleich zu Homozygoten gestiegen.
Uniparentale Disomie: Ein Phänomen, bei dem beide Kopien desselben Chromosoms von einem Elternteil vererbt werden, während das Chromosom des anderen Elternteils fehlt. Es kann als scheinbar sporadischer Fall auftreten10).
Mehr als 100 krankheitsverursachende Gene für die Retinitis pigmentosa wurden identifiziert, und selbst derselbe Phänotyp kann durch viele verschiedene Gene verursacht werden2). Diese genetische Vielfalt erschwert die Diagnose.
4. Praktische Aspekte der genetischen Beratung und der genetischen Testung
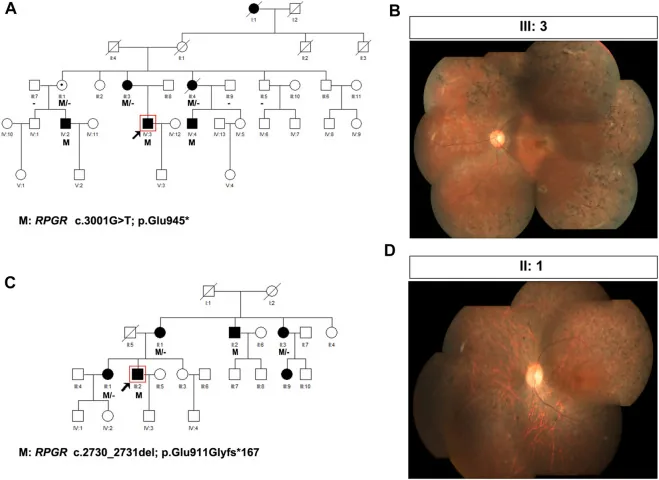
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Stammbäume und Fundusaufnahmen von zwei Familien mit X-chromosomaler Retinitis pigmentosa (RPGR-Varianten: c.3001G>T und c.2730_2731del). Die Stammbäume verwenden Standardsymbole (ausgefüllte Quadrate für betroffene Männer, gepunktete Kreise für Trägerinnen), um das Vererbungsmuster darzustellen; die entsprechenden Fundusaufnahmen zeigen eine fortgeschrittene knochenbälkchenartige Pigmentierung und Netzhautatrophie. Dies entspricht der Erstellung des Stammbaums (Pedigree) und der Bestimmung des Vererbungsmusters, die im Abschnitt „4. Praktische genetische Beratung und genetische Tests“ behandelt werden.
Für eine angemessene genetische Beratung ist folgende Vorbereitung erforderlich.
Erstellung eines Stammbaums (Pedigree): Eine Familienanamnese über mindestens drei Generationen erheben und als Stammbaum festhalten. Aus der vertikalen und horizontalen Verteilung der Betroffenen lässt sich das Vererbungsmuster abschätzen.
Abschätzung des Vererbungsmusters: Anhand von Stammbaum und Phänotyp bestimmen, ob es sich um autosomal-dominant, autosomal-rezessiv, X-chromosomal oder maternale Vererbung handelt.
Aufklärung über den Gentest und Einholung der schriftlichen Einwilligung: Bevor die genetischen Informationen des Patienten untersucht werden, müssen Bedeutung und Probleme des Tests ausreichend erklärt und eine schriftliche Einwilligung eingeholt werden.
Anonymisierung der DNA-Testergebnisse und Informationsmanagement: Die Privatsphäre durch eine rückverfolgbare Anonymisierung schützen.
Vorfestlegung des Umgangs mit Zufallsbefunden: Vor der Untersuchung mit dem Patienten klären, wie bei schwerwiegenden Zufallsbefunden mit möglicher Lebensgefahr vorzugehen ist.
Hohe Genauigkeit. Geeignet zur Bestätigung bekannter Mutationsstellen
Panel-Test (Targeted Sequencing)
Genengruppe, die mit bestimmten Krankheiten verbunden ist
Gleichzeitige Untersuchung von Genen, die mit Netzhauterkrankungen zusammenhängen. Hohe diagnostische Rate5)
Exom-Analyse (NGS)
Gesamte Exon-Regionen
Erkennt unbekannte Varianten. Nützlich, wenn es viele krankheitsverursachende Gene gibt
Gesamtgenom-Analyse (WGS)
Gesamtes Genom
Kann eine höhere diagnostische Rate haben als bestehende gezielte NGS-Tests13)
Bei hereditärer Retinadystrophie kann, wenn eine durch eine RPE65-Genmutation verursachte Erkrankung vermutet wird und etwa zur Unterstützung der Beurteilung einer Indikation für eine Gentherapie, ein Teil der Untersuchungen unter den Voraussetzungen als Kassenleistung durchgeführt werden4).
Interpretation von Varianten und wichtige Datenbanken
Die Interpretation von Varianten erfordert sorgfältige Beurteilung. Es ist bekannt, dass etwa 30 % der in Artikeln als „Varianten“ veröffentlichten Beschreibungen Polymorphismen (normale Varianten) sind. Allgemein sollten Sequenzveränderungen, die bei 1 oder mehr von 100 gesunden Personen auftreten, als Polymorphismen behandelt werden.
Folgende wichtige Datenbanken werden verwendet.
OMIM (Online Mendelian Inheritance in Man): umfassende Datenbank für Erbkrankheiten und Gene
GeneReviews: bietet Informationen zur genetischen Beratung für jede Erkrankung
RetNet (Retinal Information Network): auf Netzhauterkrankungen spezialisierte Gendatenbank
QWerden die Kosten für Gentests von der Versicherung übernommen?
A
Bei einigen erblichen Augenerkrankungen können Gentests als Kassenleistung durchgeführt werden, wenn die Indikation für eine Behandlung festgelegt wird und die Kriterien der vorgesehenen Einrichtung erfüllt sind. Ob eine Übernahme erfolgt, hängt jedoch von der Art des Tests und der zugrunde liegenden Erkrankung ab; daher ist eine Rücksprache in der behandelnden Einrichtung erforderlich. Nicht versicherte Tests können als Selbstzahlerleistung anfallen. Bei als schwer behandelbar eingestuften Erkrankungen (z. B. Retinitis pigmentosa) kann auch finanzielle Unterstützung über das Förderprogramm für medizinische Kosten bei bestimmten schwer behandelbaren Erkrankungen möglich sein4).
5. System der genetischen Beratung und Zukunft der Behandlung
Bei der Durchführung genetischer Beratung gilt die Zusammenarbeit mit einem Facharzt für Genetik und einem zertifizierten genetischen Berater als ideal. In Universitätskliniken kann eine Prüfung durch ein Ethikkomitee erforderlich sein. Da genetische Informationen nicht nur den Patienten selbst, sondern auch Familienmitglieder betreffen können, ist im Umgang damit besondere Sorgfalt erforderlich. Auch die gesellschaftliche Diskussion über den Schutz vor ungerechter Diskriminierung aufgrund genetischer Informationen schreitet voran8).
Anerkannte Seltene Krankheiten und medizinische Kostenhilfe
Bei Patientinnen und Patienten mit Seltenen Krankheiten wird der Eigenanteil an den medizinischen Kosten reduziert. Für die Summe aus ambulanter Behandlung, stationärem Aufenthalt und Arzneimitteln ist eine monatliche Obergrenze festgelegt, und es gelten einkommensabhängige Stufen.
Das Gentherapeutikum voretigene neparvovec für die durch RPE65-Genmutationen verursachte erbliche Netzhautdystrophie ist ein Präparat zur subretinalen Gabe unter Verwendung eines AAV2-Vektors. Wirksamkeit und Sicherheit wurden in randomisierten kontrollierten Studien bestätigt6). Für die Entscheidung über die Behandlungsindikation muss der Krankheitstyp durch genetische Untersuchung bestätigt werden.
Zur Antisense-Oligonukleotid-(ASO)-Therapie wird die intravitreale Gabe eines Präparats für die durch CEP290-Mutationen verursachte Leber-Kongenital-Amaurose Typ 10 (LCA10) in klinischen Studien untersucht9).
Die autologe Transplantation von retinalen Pigmentepithelzellen mithilfe von iPS-Zellen wird als regenerative Medizin für die altersabhängige Makuladegeneration erforscht7). Obwohl es sich um ein anderes Feld als die erblichen Netzhauterkrankungen handelt, steht es als reales Beispiel einer Netzhaut-Zelltherapie im Fokus.
QIst eine Gentherapie jetzt verfügbar?
A
Bei der erblichen Netzhautdystrophie durch RPE65-Mutation wurden Wirksamkeit und Sicherheit von voretigene neparvovec in RCTs6) gezeigt. Um zu bestätigen, ob die Behandlung geeignet ist, muss der Krankheitstyp durch einen Gentest bestimmt werden. Bei LCA10 mit CEP290-Mutation wird die intravitreale Gabe von ASO-Präparaten in klinischen Studien untersucht9).
Autosomal-dominante, autosomal-rezessive und X-chromosomale Vererbung folgen den Mendelschen Regeln. Allerdings können die folgenden Faktoren einfache Vorhersagen erschweren.
Penetranz: Auch wenn eine Person die Mutation trägt, erkranken nicht alle. Bei niedriger Penetranz kann das Merkmal Generationen überspringen, sodass sich das Vererbungsmuster aus dem Stammbaum schwer ableiten lässt.
Expressivität: Selbst innerhalb einer Familie mit demselben mutierten Gen kann die Schwere der Symptome unterschiedlich sein.
Gain-of-function-Mutation: Ein Mechanismus, bei dem das mutierte Protein eine neue schädliche Funktion erwirbt. Dies unterscheidet sich vom üblichen dominant-negativen Effekt.
Mitochondrien befinden sich im Zytoplasma, und nur mtDNA aus der Eizelle der Mutter wird an das Kind weitergegeben. In jeder Zelle gibt es Tausende von mtDNA-Kopien, und ein Zustand, in dem mutierte mtDNA und normale mtDNA nebeneinander vorkommen (Heteroplasmie), kann auftreten. Je höher der Anteil der Heteroplasmie, desto schwerer sind die Symptome in der Regel. Bei der Leber’schen hereditären Optikusneuropathie (LHON) machen drei Varianten—11778 (am häufigsten), 3460 und 14484—etwa 90 % aller Varianten aus.
Uniparentale Disomie ist ein Zustand, bei dem beide Chromosomen eines Paares von demselben Elternteil stammen und kein Chromosom vom anderen Elternteil vorhanden ist. Ein Kind, das von einem Träger einer autosomal-rezessiven Erkrankung geboren wird, kann erkranken, auch wenn der andere Elternteil kein Träger ist, wodurch scheinbar ein sporadischer Fall entsteht10). Neben De-novo-Varianten und compound heterozygotes sollte auch ohne Familienanamnese an eine erbliche Erkrankung gedacht werden.
Bei LCA10 mit CEP290-Varianten wird die Spleißkorrektur durch ASO-Therapie in klinischen Studien untersucht9).
Der Kandidat EDIT-101 für eine Gen-Editing-Therapie unter Verwendung von CRISPR/Cas9 wurde für LCA10 (CEP290-Varianten) in der präklinischen Entwicklung beschrieben11).
Die Präimplantationsdiagnostik (PGT-M) kann bei autosomal-dominanten und -rezessiven erblichen Erkrankungen durchgeführt werden und kann im ethischen Rahmen erwogen werden12).
Es wurde gezeigt, dass die Ganzgenomsequenzierung (WGS) die molekulare Diagnoserate bei erblichen Netzhauterkrankungen im Vergleich zu den bisherigen Standard- Gentests erhöhen kann13).
Es werden KI-gestützte Tools zur Vorhersage der Pathogenität von Genvarianten entwickelt, und eine höhere Genauigkeit bei der Interpretation von Varianten wird erwartet.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.