Risk of posterior progression
Basic approach: Observation
Barrage laser indication: Progressive enlargement, proximity to the posterior pole
Treatment goal: Prevention of further posterior progression

Retinoschisis is a condition in which the neurosensory retina splits at the inner or outer plexiform layer. This is fundamentally different from retinal detachment, where the photoreceptor layer detaches from the retinal pigment epithelium.
| Type | Typical age and sex | Typical location | Prevalence/incidence |
|---|---|---|---|
| Age-related (acquired) retinoschisis | Age 40 and older; both sexes | Inferotemporal periphery (70%) | 7–30% of people aged 40 and older |
| X-linked (congenital) retinoschisis | School age; almost exclusively males | Fovea (nearly all cases) + periphery (about half) | 1 in 5,000 to 25,000 people2) |
| Myopic foveoschisis | Middle-aged and older; high myopia | Posterior pole, macula | A certain proportion of highly myopic eyes |
Retinoschisis is a condition in which splitting occurs within the layers of the neurosensory retina, and is completely different from retinal detachment (where the full-thickness retina detaches from the retinal pigment epithelium). In retinoschisis, the connection with the retinal pigment epithelium is maintained, so the visual prognosis is generally better than that of retinal detachment. However, if holes develop in both the inner and outer layers, it may progress to retinal detachment.
Age-related retinoschisis is a condition in which physiological cystoid degeneration (Blessig-Iwanoff cysts) of the adult peripheral retina fuses and expands, leading to splitting in the outer plexiform layer or inner nuclear layer.
The prevalence is reported to be 1.65–7%2), more common in individuals aged 40 and older, and bilateral in approximately 70% of cases. It occurs preferentially in the inferotemporal peripheral retina in 70% of cases, and the elevated surface is smooth. It is not hereditary and develops with aging.
Secondary retinoschisis can be caused by proliferative membranes, vitreous traction, cystic changes, intraretinal hemorrhage, exudation, or inflammation. Specific underlying diseases include diabetic retinopathy, old retinal detachment, age-related macular degeneration, retinopathy of prematurity (ROP), and Coats disease.
Most cases are asymptomatic, and progression to the posterior pole is rare, with vision often normal. When the lesion extends beyond the equator, subjective symptoms such as visual field defects may occur.
| Finding | Characteristic | Significance for Differential Diagnosis |
|---|---|---|
| Dome-shaped elevation | Smooth, transparent hemispherical; fixed and immobile | Does not move with position change |
| Water silk appearance | Undulating luster of the inner layer | Characteristic of this condition |
| snowflakes (snowflake opacities) | yellow-white granular opacities on the inner surface of the separated layer | indicator of degeneration |
Even if an outer layer hole occurs and causes outer layer retinal detachment, the risk is low. If inner and outer holes develop and progress to detachment, surgical treatment for typical rhegmatogenous retinal detachment is required.
The main pathology is the fusion and enlargement of cystoid degeneration of Henle fibers in the outer plexiform layer due to aging. The mechanism of absolute scotoma is that separation between layers in the outer plexiform layer causes tissue disruption at synaptic junctions, blocking signal transmission of light stimuli1).
Risk factors include aging and hyperopia2). No genetic predisposition has been identified2).
OCT diagnosis plays a central role2). It can depict the extent, depth, and columnar structures (vertical tissue bridges) within the separation cavity, directly confirming the split at the outer plexiform layer.
Differentiation from retinal detachment is most important.
| Feature | Retinoschisis | Retinal detachment |
|---|---|---|
| Mobility of elevation | Fixed, immobile | Moves with position change |
| Transparency | High (clear) | Low (cloudy) |
| Nature of scotoma | Absolute scotoma | Relative scotoma |
| Surface appearance | Smooth (water silk) | Irregular, undulating |
In principle, observation is recommended. The risk of progression to retinal detachment is low, approximately 0.05% per year, and most cases are asymptomatic and stable.
Risk of posterior progression
Basic approach: Observation
Barrage laser indication: Progressive enlargement, proximity to the posterior pole
Treatment goal: Prevention of further posterior progression
Outer layer hole formation
Basic policy: Observation
Note: Outer layer breaks increase the risk of detachment
Carefully evaluate indications: Caution is needed if an inner layer hole is also present
Schisis detachment
Basic policy: Surgery according to standard rhegmatogenous retinal detachment
Surgical indications: When holes occur in both inner and outer layers and detachment progresses
Prognosis: Generally favorable
Cases with macular involvement
Basic policy: Consider vitrectomy
Surgical technique: ILM peeling + gas tamponade
Evidence: Case report level1)
Interlaminar separation in the outer plexiform layer causes tissue disruption at synaptic junctions, resulting in absolute scotoma 1). It is thought that Henle fibers are pulled apart by the fusion of age-related cystoid degeneration, leading to separation. Three-dimensional evaluation with SD-OCT has enabled precise assessment of the extent of the separation cavity and thinning of the inner and outer layers, advancing the identification of long-term natural history and progression predictors 2).
In age-related retinoschisis, interlaminar separation occurs in the outer plexiform layer. This layer contains synaptic connections between photoreceptors and ganglion cells; when separation disrupts these synapses, light stimuli cannot be transmitted to bipolar cells and ganglion cells 1). Therefore, in the separated area, light does not conduct electrical signals, and it is perceived as an absolute scotoma.
Most cases of age-related retinoschisis follow a stable course. The risk of progression to retinal detachment is extremely low, approximately 0.05% per year. However, if holes develop in both the inner and outer layers, the risk of detachment increases, so regular fundus examination follow-up is important 2).
X-linked retinoschisis (XLRS) is a congenital retinal disease with X-linked recessive inheritance caused by mutations in the RS1 gene. It is a relatively rare vitreoretinal degenerative disease, with an estimated prevalence of 1 in 5,000 to 25,000 individuals 2). Typically, only males are affected, while females are carriers.
Usually only males are affected. Because it is an X-linked recessive inheritance, females with a mutation on only one X chromosome are carriers and are usually asymptomatic. Rarely, skewed X-inactivation or uniparental disomy has been reported to cause mild phenotypes in females2).
Symptoms:
Clinical findings:
Macular findings
Foveal schisis (almost all cases): Cystoid changes with spoke-wheel folds (cysts and bridging structures on OCT).
Macular atrophy: Cystoid changes regress with age and transition to atrophy; often appears similar to simple cystoid macular edema
Fluorescein angiography: Unlike cystoid macular edema, no fluorescein leakage (fovea is normal or shows window defect)
Peripheral Findings
Peripheral retinoschisis (about half): Predilection for inferotemporal quadrant; may be associated with large inner retinal holes
Gold foil reflex: Characteristic finding in the peripheral retina; accompanied by white sheathing of retinal vessels
Vitreous veil: Seen in severe cases; vitreous is not detached and adheres strongly to the retina
Complications: Retinal detachment (5–20%), vitreous hemorrhage, intra-schisis cavity hemorrhage
Suspect this disease when bilateral cystoid macular edema is observed in male children is an important diagnostic clue.
Differential diagnosis:
| Disease | Key differentiating points |
|---|---|
| Rhegmatogenous retinal detachment | Usually unilateral; demarcation line formation; associated with lattice degeneration |
| Cystoid macular edema | Fluorescein angiography shows petaloid leakage; no leakage in XLRS |
| Age-related (degenerative) retinoschisis | Occurs in elderly; limited to periphery; normal b-wave |
| Goldman-Favre syndrome | With night blindness and pigmentation |
In cystoid macular edema (CME), fluorescein angiography shows petaloid leakage and pooling of fluorescein. In XLRS, although a foveal schisis cavity exists, the blood-retinal barrier is normal, and fluorescein angiography shows no leakage, only window defects. This difference is important in differential diagnosis combining OCT and fluorescein angiography.
There is no established curative treatment.
Pharmacotherapy:
Surgical treatment:
Patient management:
Cystoid changes in the fovea may naturally decrease with growth 2). However, some cases progress to macular atrophy, and regression does not necessarily mean improvement in visual acuity. In some cases, peripheral schisis may decrease after the occurrence of posterior vitreous detachment.
Retinoschisin, encoded by the RS1 gene, is a secreted protein of 224 amino acids that forms homo-oligomeric complexes via its discoidin domain 2). By binding to photoreceptors and bipolar cells, it maintains the structural and synaptic integrity of the retina. Disruption of these complexes due to RS1 mutations leads to characteristic splitting cavities and impaired retinal signal transmission 2).
Histopathologically, retinal splitting occurs mainly in the nerve fiber layer. OCT shows splitting in the outer plexiform layer and inner nuclear layer at and around the fovea. Even when morphological splitting is localized, electrophysiology reveals global retinal dysfunction, as indicated by a negative b-wave.
Genotype-phenotype correlation (Korean cohort of 83 patients, Lee et al. 2025) 2):
In a study analyzing the schisis cavity fluid in XLRS, interphotoreceptor retinoid-binding protein (IRBP) was identified in the fluid, suggesting possible retinal metabolic abnormalities within the schisis cavity 2). Additionally, cases of exudative maculopathy, which is not typically seen in XLRS, have been reported, indicating the presence of diverse phenotypes under the same diagnosis 2).
RS1 gene therapy clinical trials are ongoing2)3). Animal models and early clinical trials have reported partial recovery of electroretinogram responses and improvement in retinal anatomy.
Key clinical trials:
Current challenges: In XLRS, retinal fragility is increased, making posterior vitreous detachment and subretinal injection technically very difficult3). Intravitreal administration may be preferable, but the risk of vitritis must be tolerated.
Gene therapy for XLRS is still in clinical trials and has not been approved as a treatment in general hospitals. Some clinical trials have revealed safety issues such as intraocular inflammation, and some trials have been discontinued 3). If you wish to participate, consultation with a specialized facility is necessary.
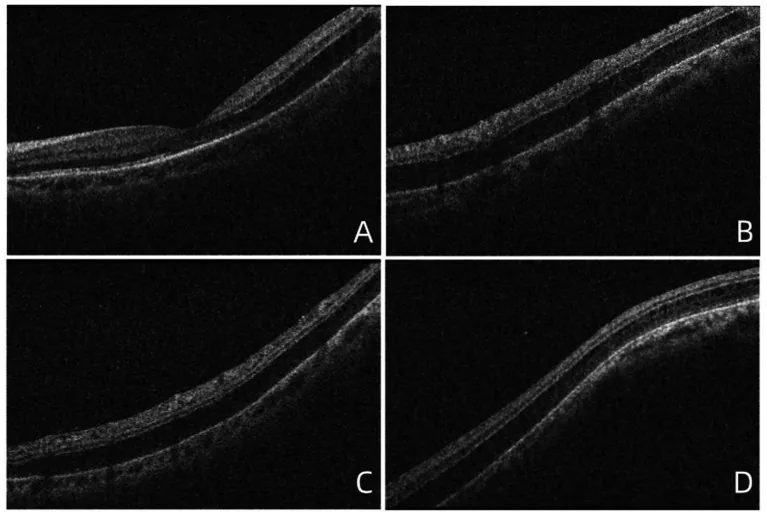
Myopic foveoschisis is a separation of the inner retinal layers in the posterior pole associated with high myopia. It has long been described as a posterior pole retinal detachment associated with high myopia, and currently the concept includes cases that also have a macular hole.
The pathophysiology involves a combination of the following factors:
Symptoms: Decreased visual acuity, central scotoma, metamorphopsia, and blurred vision. In high myopia, symptoms may be subtle.
Diagnosis (OCT):
Treatment: Vitrectomy is the basic approach.
Prognosis: