Epidermolysis Bullosa Simplex (EBS)
Number of subtypes: 14
Inheritance: Autosomal dominant
Lesion site: Intraepidermal (basal cell layer)

Epidermolysis bullosa (EB) is a group of hereditary skin diseases characterized by dysfunction of the dermal-epidermal junction. Due to abnormalities in structural proteins of the cutaneous basement membrane zone, blisters form even with minor mechanical trauma.
There are 34 subtypes, classified into 4 major groups.
Epidermolysis Bullosa Simplex (EBS)
Number of subtypes: 14
Inheritance: Autosomal dominant
Lesion site: Intraepidermal (basal cell layer)
Junctional EB (JEB)
Number of subtypes: 9
Inheritance: Autosomal recessive
Lesion site: Lamina lucida
Dystrophic EB (DEB)
Number of subtypes: 11
Inheritance: Both autosomal dominant and recessive
Lesion site: Sublamina densa (deficiency or reduction of anchoring fibrils)
Kindler EB
Reported cases: Approximately 250 worldwide
Inheritance: Autosomal recessive
Lesion site: Involves multiple layers
Epidermolysis bullosa acquisita (EBA) is an autoimmune disease that differs from the above, typically developing in the 30s to 40s. It is caused by autoantibodies against type VII collagen.
More than 16 causative genes have been identified, and mutations lead to abnormalities in structural proteins of the epidermis, basement membrane, and upper dermis. Severity depends on the type and location of the gene mutation.
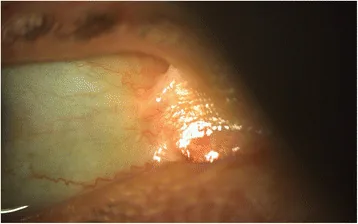
Ocular involvement can occur in all subtypes but is more frequent and severe in recessive dystrophic, junctional, Kindler, and severe simplex types. Onset can be as early as 1 month of age.
Various lesions occur in the cornea, eyelids, conjunctiva, and lacrimal ducts.
| Site | Main Findings |
|---|---|
| Cornea | Erosion, scarring, neovascularization |
| Eyelid | Ectropion, entropion, blister formation |
| Conjunctiva | Symblepharon, cicatricial changes |
Corneal lesions are the most frequent. Recurrent corneal erosion and abrasions occur repeatedly, progressing to corneal scarring and neovascularization. Ultimately, it may lead to limbal stem cell deficiency or refractory epithelial defects.
Eyelid ectropion results from blister formation and cicatricial contracture around the eyelids. When combined with entropion, it causes trichiasis and worsens corneal epithelial damage.
Symblepharon forms when epithelial defects of the bulbar and palpebral conjunctiva persist, accompanied by inflammatory reactions. Progression leads to restricted eye movement and incomplete eyelid closure.
In the dystrophic type, the following complications have also been reported:
Ocular complications can also cause refractive errors, strabismus, and amblyopia.
Corneal abnormalities (recurrent corneal erosion, abrasion, scarring, neovascularization) are the most common, followed by ectropion and eyelid blister formation. These can occur in all subtypes, but are particularly severe in recessive dystrophic and junctional types.
Epidermolysis bullosa is caused by mutations in genes encoding structural proteins of the cutaneous basement membrane zone.
All patients with epidermolysis bullosa should be referred to an ophthalmologist for baseline examination. Thereafter, regular follow-up should be performed according to disease severity.
Management of epidermolysis bullosa requires multidisciplinary collaboration including dermatology, pediatrics, ophthalmology, and plastic surgery. Since ocular complications can appear from infancy, prompt detection and treatment of vision-threatening lesions are important.
Preservative-free lubricants (artificial tears, eye ointments) to maintain ocular surface moisture are the mainstay of treatment. This is the most important intervention for preventing corneal and mucosal erosions.
For recurrent corneal erosions, antibiotic eye drops are used to prevent infection and hyaluronic acid eye drops to promote epithelial repair. The use of therapeutic soft contact lenses and application of eye ointment before bedtime are also effective.
When corneal lesions or symblepharon progress, the following surgical interventions are considered.
For entropion and ectropion, surgical intervention is the main treatment1).
For ectropion, the following surgical techniques are selected according to severity.
For trichiasis associated with entropion, symptomatic treatment by epilation is performed, but if recurrence is frequent, electrolysis or eyelash relocation (Machek procedure, Spencer-Watson procedure) is considered.
For symblepharon, consider Z-plasty of the adhesive tissue or conjunctival sac reconstruction using conjunctival or amniotic membrane transplantation. However, if tear secretion is severely impaired, mucosal transplantation is often ineffective.
Symptomatic treatment is the mainstay of disease management, with avoidance of trauma, appropriate wound care, and disinfection as the basics.
The following systemic drug therapies have shown some efficacy in small-scale studies.
Eyelid scrubs (cleaning/washing of the eyelids) should not be used in patients with epidermolysis bullosa because they can cause blisters on the eyelids. Eyelid hygiene should be maintained using more gentle methods.
Wearing glasses is possible, but blisters may occur at the contact points of the frame. It is necessary to choose lightweight frames with a wide contact surface and minimal pressure, and to use cushioning materials for the nose pads.
The cutaneous basement membrane zone is a structure that firmly adheres the epidermis and dermis, and is composed of the following layers.
In simplex EB, keratin filaments within basal epidermal cells are fragile, leading to blister formation at the most superficial layer. In junctional EB, deficiency or reduction of laminin 332 or type XVII collagen causes tissue separation in the lamina lucida. In dystrophic EB, abnormalities in type VII collagen lead to deficiency or reduction of anchoring fibrils, forming clefts below the lamina densa. Kindler syndrome presents with lesions spanning multiple layers.
The periorbital area and ocular surface are subject to the same shear forces and blister formation as the skin. Because the conjunctival and corneal epithelium also have a basement membrane structure, epithelial detachment, erosion, and blistering occur through the same mechanism as skin lesions.
Repeated damage to the corneal epithelium leads to chronic poor adhesion of the corneal epithelial basement membrane, forming the pathology of recurrent corneal epithelial erosion. In the eyelids, repeated blister formation and scar contracture progress to ectropion, entropion, and trichiasis. Persistent conjunctival epithelial defects lead to symblepharon, resulting in conjunctival sac shortening and impaired eye movement.
Gene therapy aimed at fundamentally modifying the disease activity of epidermolysis bullosa is the most promising area of research. The goal is to restore normal production of structural proteins by correcting or supplementing the abnormal gene.
Losartan (ARB) has been reported as a potential first-line drug to delay fibrotic changes in recessive dystrophic epidermolysis bullosa. It is expected to have anti-fibrotic effects through inhibition of TGF-β signaling.
- TFOS DEWS III: Management and Therapy of Dry Eye Disease. Am J Ophthalmol. 2025.