Epidermolysis bullosa simplex (EBS)
Anzahl der Subtypen : 14
Vererbungsmodus : Autosomal-dominant
Läsionsort : Intraepidermal (Basalzellschicht)

Epidermolysis bullosa (EB) ist eine Gruppe erblicher Hauterkrankungen, die durch eine Funktionsstörung der dermoepidermalen Junktion gekennzeichnet sind. Aufgrund von Anomalien der Strukturproteine der Basalmembranzone der Haut kommt es bereits bei geringfügigem mechanischem Trauma zur Blasenbildung.
Es gibt 34 Subtypen, die in 4 Hauptgruppen eingeteilt werden.
Epidermolysis bullosa simplex (EBS)
Anzahl der Subtypen : 14
Vererbungsmodus : Autosomal-dominant
Läsionsort : Intraepidermal (Basalzellschicht)
Junktionale EB (JEB)
Anzahl der Subtypen : 9
Vererbungsmodus : Autosomal-rezessiv
Läsionsort : Lamina lucida
Dystrophe EB (DEB)
Anzahl der Subtypen : 11
Vererbungsmodus : Autosomal-dominant und -rezessiv
Läsionsort : Unterhalb der Lamina densa (Mangel oder Fehlen von Ankerfibrillen)
Kindler-EB
Berichtete Fälle : Weltweit etwa 250
Vererbungsmodus : Autosomal-rezessiv
Läsionsort : Betrifft mehrere Schichten
Die erworbene Epidermolysis bullosa (EBA) ist eine Autoimmunerkrankung, die im Alter von 30–40 Jahren auftritt und sich von den oben genannten unterscheidet. Sie wird durch Autoantikörper gegen Kollagen Typ VII verursacht.
Es wurden mehr als 16 ursächliche Gene identifiziert; Mutationen führen zu Anomalien der Strukturproteine der Epidermis, Basalmembran und oberen Dermis. Der Schweregrad hängt von der Art und Lage der Genmutation ab.
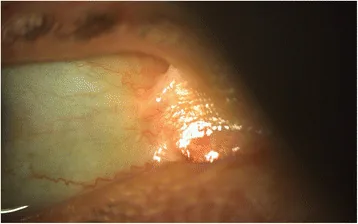
Augenläsionen können bei allen Subtypen auftreten, sind jedoch bei rezessiv-dystrophen, junktionalen, Kindler- und schweren einfachen Formen häufiger und schwerwiegender. Der Beginn kann bereits im Alter von einem Monat beobachtet werden.
An Hornhaut, Augenlidern, Bindehaut und Tränenwegen treten vielfältige Läsionen auf.
| Lokalisation | Hauptbefunde |
|---|---|
| Hornhaut | Erosion, Narbe, Neovaskularisation |
| Augenlid | Ektropium, Entropium, Blasenbildung |
| Bindehaut | Symblepharon, narbige Veränderungen |
Hornhautläsionen sind am häufigsten. Wiederkehrende Hornhauterosionen und -abschürfungen treten auf und entwickeln sich zu Hornhautnarben und Neovaskularisation. Dies kann schließlich zu einer Limbusstammzellinsuffizienz oder einem therapierefraktären Epitheldefekt führen.
Das Ektropium des Augenlids entsteht durch Blasenbildung und narbige Schrumpfung um die Augenlider. Wenn es mit einem Entropium einhergeht, verursacht es Trichiasis, die die Hornhautepithelschädigung verschlimmert.
Das Symblepharon entsteht, wenn Epitheldefekte der bulbären und tarsalen Bindehaut bestehen bleiben und eine Entzündungsreaktion hinzukommt. Im fortgeschrittenen Stadium kann es zu Bewegungseinschränkungen des Auges und unvollständigem Lidschluss führen.
Beim dystrophen Typ wurden auch folgende Komplikationen berichtet:
Augenkomplikationen können auch zu Refraktionsfehlern, Strabismus und Amblyopie führen.
Hornhautanomalien (rezidivierende Hornhauterosionen, Abschürfungen, Narbenbildung, Neovaskularisation) sind am häufigsten, gefolgt von Ektropium und Blasenbildung der Augenlider. Alle Subtypen können betroffen sein, aber die dystrophe recessive und die junktionale Form sind besonders schwerwiegend.
Epidermolysis bullosa wird durch Mutationen in Genen verursacht, die für Strukturproteine der Basalmembranzone der Haut kodieren.
Alle Patienten mit Epidermolysis bullosa sollten zur Basisuntersuchung an einen Augenarzt überwiesen werden. Danach sollte je nach Schweregrad der Erkrankung eine regelmäßige Nachsorge erfolgen.
Das Management der Epidermolysis bullosa erfordert eine multidisziplinäre Zusammenarbeit, einschließlich Dermatologie, Pädiatrie, Augenheilkunde und plastischer Chirurgie. Augenkomplikationen können bereits im Säuglingsalter auftreten, daher ist eine schnelle Erkennung und Behandlung sehbedrohender Läsionen wichtig.
Die Aufrechterhaltung der Befeuchtung der Augenoberfläche mit konservierungsmittelfreien Gleitmitteln (künstliche Tränen, Augensalben) ist die Säule der Behandlung. Dies ist die wichtigste Maßnahme zur Vorbeugung von Hornhaut- und Schleimhauterosionen.
Bei rezidivierenden Hornhauterosionen werden antibiotische Augentropfen zur Infektionsprophylaxe und Hyaluronsäure-Augentropfen zur Förderung der Epithelheilung eingesetzt. Auch die Verwendung von therapeutischen weichen Kontaktlinsen und das Auftragen von Augensalbe vor dem Schlafengehen sind wirksam.
Bei Fortschreiten von Hornhautläsionen oder Symblepharon werden die folgenden chirurgischen Eingriffe in Betracht gezogen.
Bei Entropium und Ektropium ist der chirurgische Eingriff die Hauptbehandlung1).
Beim Ektropium werden je nach Schweregrad die folgenden Verfahren ausgewählt.
Bei Trichiasis infolge eines Entropiums wird eine symptomatische Behandlung durch Epilation durchgeführt. Bei wiederholtem Auftreten werden jedoch Elektrolyse der Wimpern oder Wimperntransposition (Machek-Verfahren oder Spencer-Watson-Verfahren) in Betracht gezogen.
Bei Symblepharon ist eine Z-Plastik des adhärenten Gewebes oder eine Rekonstruktion des Bindehautsacks mittels Bindehaut- oder Amnionmembrantransplantation in Betracht zu ziehen. Bei stark eingeschränkter Tränensekretion ist eine Schleimhauttransplantation jedoch oft wirkungslos.
Die symptomatische Behandlung ist der Hauptpfeiler des Krankheitsmanagements, wobei die Vermeidung von Traumata, angemessene Wundversorgung und Desinfektion die Grundlage bilden.
Als systemische medikamentöse Therapie haben die folgenden Substanzen in kleinen Studien eine gewisse Wirksamkeit gezeigt.
Ein Lid-Scrub (Reinigung und Waschen der Augenlider) kann Blasen an den Augenlidern verursachen und sollte daher bei Patienten mit Epidermolysis bullosa nicht angewendet werden. Die Sauberkeit der Augenlider muss mit schonenderen Methoden erhalten werden.
Das Tragen einer Brille ist möglich, aber an den Kontaktstellen des Gestells können Blasen auftreten. Es ist wichtig, ein leichtes Gestell mit großer Auflagefläche und geringem Druck zu wählen und an den Nasenpads Polstermaterial zu verwenden.
Die Hautbasalmembranzone ist eine Struktur, die die Epidermis fest mit der Dermis verbindet und aus den folgenden Schichten besteht.
Bei der einfachen EB sind die Keratinfilamente in den basalen Epidermiszellen brüchig, und es bilden sich Blasen in der oberflächlichsten Schicht. Bei der junktionalen EB führt ein Mangel oder eine Verminderung von Laminin 332 oder Kollagen Typ XVII zu einer Gewebetrennung in der Lamina lucida. Bei der dystrophen EB führt eine Anomalie des Kollagens Typ VII zu einem Mangel oder einer Verminderung der Verankerungsfibrillen, und es bilden sich Spalten unter der Lamina densa. Der Kindler-Typ zeigt Läsionen, die mehrere Schichten betreffen.
Die periorbitale Region und die Augenoberfläche sind denselben Scherkräften und Blasenbildungen ausgesetzt wie die Haut. Da auch das Epithel der Bindehaut und Hornhaut eine Basalmembranstruktur aufweist, kommt es durch denselben Mechanismus wie bei den generalisierten Hautläsionen zu Epithelablösungen, Erosionen und Blasen.
Wiederholte Schädigungen des Hornhautepithels führen zu einer Chronifizierung der schlechten Adhäsion der Hornhautepithel-Basalmembran und bilden das Krankheitsbild der rezidivierenden Hornhautepithel-Erosion. An den Augenlidern führt die wiederholte Blasenbildung und Narbenkontraktur zu Ektropium, Entropium und Trichiasis. Anhaltende Bindehautepitheldefekte führen zu einer Symblepharon, die eine Verkürzung des Bindehautsacks und eine Einschränkung der Augenbewegungen verursacht.
Die Gentherapie, die darauf abzielt, den Krankheitsverlauf der Epidermolysis bullosa grundlegend zu verändern, ist das vielversprechendste Forschungsgebiet. Ziel ist es, durch Ansätze zur Korrektur oder Ergänzung des defekten Gens die normale Produktion von Strukturproteinen wiederherzustellen.
Losartan (ARB) könnte bei rezessiver dystrophischer Epidermolysis bullosa ein Mittel der ersten Wahl sein, um fibrotische Veränderungen zu verlangsamen. Es wird eine antifibrotische Wirkung durch Hemmung des TGF-β-Signalwegs erwartet.
- TFOS DEWS III: Management and Therapy of Dry Eye Disease. Am J Ophthalmol. 2025.