Risque de progression postérieure
Principe de base : Surveillance
Indication du laser barrière : Extension progressive, proximité du pôle postérieur
Objectif du traitement : Empêcher la progression vers l’arrière

Le rétinoschisis est une affection dans laquelle la rétine neurosensorielle se sépare au niveau de la couche plexiforme interne ou externe. Ce mécanisme est fondamentalement différent de celui du décollement de la rétine, où la couche des photorécepteurs se détache de l’épithélium pigmentaire.
| Forme clinique | Âge et sexe de prédilection | Site de prédilection | Prévalence / Incidence |
|---|---|---|---|
| Rétinoschisis lié à l’âge (acquis) | À partir de 40 ans ; hommes et femmes | Périphérie inféro-temporale (70 %) | 7 à 30 % des personnes de plus de 40 ans |
| Rétinoschisis lié à l’X (congénital) | Âge scolaire ; presque exclusivement masculin | Fovéa (presque tous les cas) + périphérie (environ la moitié) | 1 personne sur 5 000 à 25 0002) |
| Rétinoschisis myopique fovéal | Après l’âge moyen ; forte myopie | Pôle postérieur et macula | Une certaine proportion des yeux fortement myopes |
Le rétinoschisis est une affection où une séparation se produit « à l’intérieur des couches » de la rétine neurosensorielle, ce qui est totalement différent du décollement de la rétine (où toute l’épaisseur de la rétine se détache de l’épithélium pigmentaire). Dans le rétinoschisis, la connexion avec l’épithélium pigmentaire rétinien est maintenue, de sorte que le pronostic visuel est généralement meilleur que dans le décollement de la rétine. Cependant, si des trous se forment dans les deux couches interne et externe, il existe un risque de progression vers un décollement de la rétine.
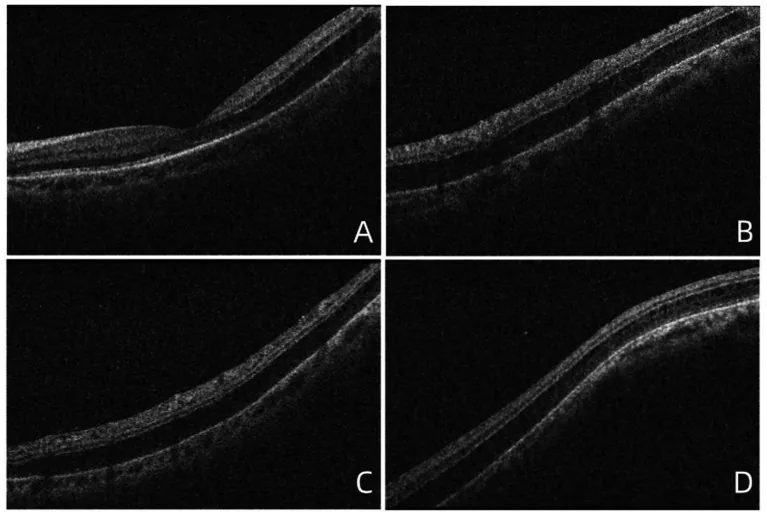
La rétinoschisis liée à l’âge est une condition pathologique dans laquelle la dégénérescence kystique physiologique de la rétine périphérique adulte (kystes de Blessig-Iwanoff) fusionne et s’élargit, entraînant une séparation dans la couche plexiforme externe ou la couche granulaire interne.
La prévalence est de 1,65 à 7 %2), plus fréquente après 40 ans, et bilatérale dans environ 70 % des cas. 70 % surviennent dans la région périphérique inféro-temporale de la rétine, avec une surface surélevée lisse. Il n’y a pas de caractère héréditaire et la maladie se développe avec l’âge.
Le rétinoschisis secondaire peut être causé par une membrane proliférative, une traction vitréenne, des modifications kystiques, une hémorragie intrarétinienne, un exsudat ou une inflammation. Les causes spécifiques incluent la rétinopathie diabétique, le décollement de rétine ancien, la dégénérescence maculaire liée à l’âge, la rétinopathie du prématuré (ROP) et la maladie de Coats.
La plupart des cas sont asymptomatiques ; l’extension jusqu’au pôle postérieur est rare et l’acuité visuelle est souvent normale. Lorsque les lésions dépassent l’équateur, des symptômes subjectifs tels qu’un déficit du champ visuel peuvent apparaître.
| Signe | Caractéristique | Importance pour le diagnostic différentiel |
|---|---|---|
| Élévation en dôme | Hémisphérique lisse et transparent ; fixe et immobile | Ne se déplace pas avec les changements de position |
| Aspect de soie mouillée | Reflet ondulé de la couche interne | Caractéristique de cette affection |
| snowflakes (opacités en flocons de neige) | Opacités granuleuses jaune-blanc sur la face interne de la couche de séparation | Indicateur de dégénérescence |
Même si un trou externe se forme et provoque un décollement de la couche externe de la rétine, le risque est faible. Si des trous interne et externe se forment et évoluent vers un décollement, un traitement chirurgical standard pour le décollement de rétine rhegmatogène est nécessaire.
Le principal mécanisme pathologique est la fusion et l’expansion de la dégénérescence kystique des fibres de Henle dans la couche plexiforme externe due au vieillissement. Le mécanisme du scotome absolu est que la séparation lamellaire dans la couche plexiforme externe provoque une rupture tissulaire au niveau des jonctions synaptiques, interrompant ainsi la transmission du signal lumineux1).
Les facteurs de risque incluent le vieillissement et l’hypermétropie2). Aucune prédisposition génétique n’a été identifiée2).
Le diagnostic par OCT joue un rôle central2). Il permet de visualiser l’étendue, la profondeur de la cavité de séparation et les structures en colonnes entre les couches (ponts tissulaires verticaux), et de confirmer directement la séparation au niveau de la couche plexiforme externe.
La distinction avec un décollement de la rétine est primordiale.
| Caractéristique | Rétinoschisis | Décollement de la rétine |
|---|---|---|
| Mobilité du soulèvement | Fixe et immobile | Mobile avec les changements de position |
| Transparence | Élevée (transparent) | Faible (trouble) |
| Nature du scotome | Scotome absolu | Scotome relatif |
| Aspect de surface | Lisse (water silk) | Irrégulier, ondulé |
En principe, une simple surveillance est recommandée. Le risque de progression vers un décollement de la rétine est faible, d’environ 0,05 % par an, et la plupart des cas sont asymptomatiques et stables.
Risque de progression postérieure
Principe de base : Surveillance
Indication du laser barrière : Extension progressive, proximité du pôle postérieur
Objectif du traitement : Empêcher la progression vers l’arrière
Formation d'une déchirure de la couche externe
Principe de base : Surveillance
Point d’attention : Les déchirures de la couche externe augmentent le risque de décollement
Évaluer soigneusement les indications : Prudence en cas de trou dans la couche interne simultané
Décollement par rétinoschisis
Principe de base : Chirurgie similaire à celle du décollement de rétine rhegmatogène standard
Indications chirurgicales : Lorsque des trous se forment dans les deux couches interne et externe et que le décollement progresse
Pronostic : Généralement bon
Cas d'infiltration maculaire
Principe de base : Envisager une vitrectomie
Technique chirurgicale : Pelage de la limitante interne + tamponnement gazeux
Données probantes : Niveau de rapports de cas1)
La séparation lamellaire au niveau de la couche plexiforme externe provoque une rupture des jonctions synaptiques, entraînant un scotome absolu1). On pense que la séparation est causée par l’écartement des fibres de Henle dû à la fusion de dégénérescences kystiques liées à l’âge. L’évaluation tridimensionnelle par SD-OCT permet de mesurer précisément l’étendue de la cavité de séparation et la finesse des couches internes et externes, ce qui a permis d’identifier l’évolution naturelle à long terme et les facteurs prédictifs de progression2).
Dans le rétinoschisis lié à l’âge, une séparation lamellaire se produit au niveau de la couche plexiforme externe. Cette région contient les synapses entre les photorécepteurs et les cellules ganglionnaires ; lorsque la séparation rompt ces synapses, la stimulation lumineuse n’est plus transmise aux cellules bipolaires et ganglionnaires1). Ainsi, dans la zone de séparation, même en présence de lumière, le signal électrique n’est pas conduit, ce qui est perçu comme un scotome absolu.
La plupart des rétinoschisis liés à l’âge ont une évolution stable. Le risque de progression vers un décollement de la rétine est extrêmement faible, d’environ 0,05 % par an. Cependant, si des trous se forment dans les deux couches interne et externe, le risque de décollement augmente, d’où l’importance d’un suivi régulier par examen du fond d’œil 2).
Le rétinoschisis lié à l’X (XLRS) est une maladie rétinienne congénitale à transmission récessive liée à l’X due à des mutations du gène RS1. Il s’agit d’une maladie vitréo-rétinienne dégénérative rare, avec une prévalence estimée entre 1 personne sur 5 000 et 1 sur 25 000 2). Seuls les hommes sont généralement atteints, les femmes étant conductrices.
Habituellement, seuls les hommes sont atteints. En raison de l’hérédité récessive liée à l’X, les femmes porteuses d’une mutation sur un seul chromosome X sont généralement asymptomatiques. Rarement, en raison d’une inactivation déséquilibrée du chromosome X ou d’une disomie uniparentale, un phénotype léger peut apparaître chez les femmes2).
Symptômes subjectifs :
Signes cliniques :
Atteinte maculaire
Séparation fovéolaire (presque tous les cas) : modifications kystiques avec plis en rayon de roue (kystes et ponts en OCT)
Atrophie maculaire : avec l’âge, les modifications kystiques régressent et évoluent vers l’atrophie ; de nombreux cas ressemblent à un simple œdème maculaire kystique
Angiographie à la fluorescéine : contrairement à l’œdème maculaire kystique, il n’y a pas de fuite de fluorescéine (la fovéa est normale ou présente un window defect)
Signes périphériques
Schisis rétinien périphérique (environ la moitié des cas) : prédominance dans le quadrant inférotemporal ; peut s’accompagner de grands trous rétiniens internes
Reflet doré : signe caractéristique de la rétine périphérique ; accompagné d’un blanchiment des vaisseaux rétiniens
Voile vitréen : observé dans les cas graves ; le vitré n’est pas détaché et adhère fortement à la rétine
Complications : décollement de la rétine (5 à 20 %), hémorragie du vitré, hémorragie intra-kystique
Il est important de suspecter cette maladie chez un garçon présentant un œdème maculaire cystoïde bilatéral, ce qui constitue une porte d’entrée diagnostique clé.
Diagnostic différentiel :
| Maladie | Points clés du diagnostic différentiel |
|---|---|
| Décollement de rétine rhegmatogène | Généralement unilatéral ; formation de ligne de démarcation ; associé à une dégénérescence en lattice |
| Œdème maculaire cystoïde | Angiographie à la fluorescéine montre une fuite en pétales ; pas de fuite dans le XLRS |
| Rétinoschisis lié à l’âge (dégénératif) | Survient chez les personnes âgées ; limité à la périphérie ; onde b normale |
| Syndrome de Goldman-Favre | Accompagné de cécité nocturne et de dépôts pigmentaires |
Dans l’œdème maculaire cystoïde (OMC), l’angiographie à la fluorescéine montre une fuite et une accumulation de fluorescence en forme de pétale. En revanche, dans le XLRS, il existe une cavité de schisis fovéolaire mais la barrière hémato-rétinienne est normale, et l’angiographie ne montre pas de fuite, seulement un effet de fenêtre (window defect). Cette différence est importante pour le diagnostic différentiel combinant OCT et angiographie.
Il n’existe pas de traitement curatif établi.
Traitement médicamenteux :
Traitement chirurgical :
Prise en charge du patient :
Avec la croissance, les modifications kystiques de la fovéa peuvent s’atténuer spontanément2). Cependant, certains cas évoluent vers une atrophie maculaire, et la régression ne signifie pas nécessairement une amélioration de l’acuité visuelle. Dans certains cas, il a été rapporté que la schisis périphérique diminue après l’apparition d’un décollement postérieur du vitré.
La rétinoschisine codée par le gène RS1 est une protéine sécrétée de 224 acides aminés qui forme des complexes homo-oligomères via le domaine discoïdine 2). En se liant aux photorécepteurs et aux cellules bipolaires, elle maintient l’intégrité structurale et synaptique de la rétine. La rupture de ces complexes due à une mutation de RS1 entraîne la formation de cavités de schisis caractéristiques et une altération de la transmission du signal rétinien 2).
Histopathologiquement, le schisis rétinien se produit principalement dans la couche des fibres nerveuses. L’OCT montre un clivage au niveau de la couche plexiforme externe et de la couche granulaire interne au niveau de la fovéa et de ses alentours. Même si le schisis morphologique est localisé, l’électrophysiologie révèle un dysfonctionnement rétinien global, comme l’indique l’onde b négative.
Corrélation génotype-phénotype (cohorte coréenne de 83 cas, Lee et al. 2025) 2) :
Une étude analysant le liquide de cavitation du XLRS a identifié la protéine de liaison au rétinoïde inter-photorécepteurs (IRBP) dans le liquide de cavitation, suggérant une possible anomalie métabolique rétinienne dans la cavité de séparation2). De plus, des cas de maculopathie exsudative, habituellement absente dans le XLRS, ont été rapportés, indiquant l’existence de phénotypes variés sous le même diagnostic2).
Thérapie génique RS1 : des essais cliniques sont en cours2)3). Des études sur modèles animaux et des essais cliniques précoces ont rapporté une récupération partielle des réponses électrorétinographiques et une amélioration de l’anatomie rétinienne.
Principaux essais cliniques :
Défi actuel : Dans la XLRS, la fragilité rétinienne est accrue, ce qui rend le décollement de la membrane hyaloïde postérieure et l’injection sous-rétinienne techniquement très difficiles 3). L’administration intravitréenne peut être préférable dans certains cas, mais il faut accepter le risque d’uvéite.
La thérapie génique pour la XLRS est encore au stade d’essai clinique et n’est pas approuvée comme traitement dans les hôpitaux généraux. Plusieurs essais cliniques ont révélé des problèmes de sécurité tels qu’une inflammation intraoculaire, et certains essais ont été interrompus 3). Pour participer, une consultation dans un centre spécialisé est nécessaire.
Le myopic foveoschisis est une séparation des couches internes de la rétine au niveau du pôle postérieur associée à une myopie forte. Décrit historiquement comme un décollement rétinien du pôle postérieur lié à la myopie forte, il inclut désormais les cas compliqués d’un trou maculaire.
La physiopathologie implique plusieurs facteurs combinés :
Symptômes subjectifs : baisse de l’acuité visuelle, scotome central, métamorphopsie, vision trouble. Dans la myopie forte, les symptômes subjectifs peuvent être discrets.
Diagnostic (OCT) :
Traitement : La vitrectomie est la base.
Pronostic :