Arkaya İlerleme Riski
Temel prensip: Gözlem
Bariyer lazer endikasyonu: İlerleyici genişleme, arka kutba yakınlık
Tedavi amacı: Daha fazla arka ilerlemenin önlenmesi

Retinoskizis, nöral retinanın iç pleksiform veya dış pleksiform tabakada ayrılması durumudur. Fotoreseptör tabakasının pigment epitelinden ayrıldığı retina dekolmanından temel olarak farklıdır.
| Alt Tip | Sık Görülen Yaş ve Cinsiyet | Sık Görülen Bölge | Prevalans ve İnsidans |
|---|---|---|---|
| Yaşa bağlı (edinilmiş) retina şizisi | 40 yaş ve üzeri; kadın ve erkek | Alt temporal perifer (%70) | 40 yaş üzerindekilerin %7-30’u |
| X’e bağlı (konjenital) retina ayrılması | Okul çağı; neredeyse sadece erkekler | Fovea (neredeyse tüm vakalar) + perifer (yaklaşık yarısı) | 5.000 ila 25.000 kişide 1 2) |
| Miyopik foveal retinoskizis | Orta yaş ve üzeri; yüksek miyopi | Arka kutup, makula | Yüksek miyopik gözlerin belirli bir oranı |
Retinoskizis, nöral retinanın “katman içinde” ayrılmasıyla oluşan bir durumdur ve retina dekolmanından (retinanın tüm katmanlarının pigment epitelinden ayrılması) tamamen farklıdır. Retinoskiziste retina pigment epiteli ile bağlantı korunduğu için görme prognozu genellikle retina dekolmanına göre daha iyidir. Ancak iç ve dış katmanlarda delik oluşursa retina dekolmanına ilerleme olasılığı vardır.
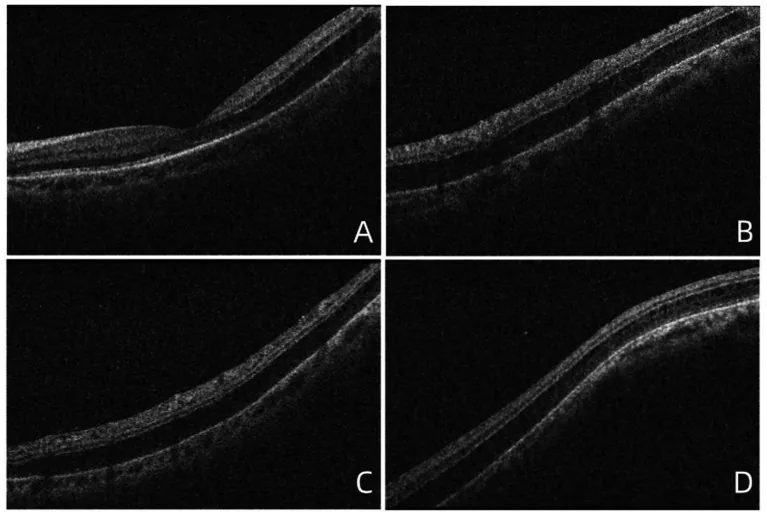
Yaşa bağlı retinoskizis, erişkin periferik retinasındaki fizyolojik kistoid dejenerasyonun (Blessig-Iwanoff kistleri) birleşip genişleyerek dış pleksiform veya iç granüler tabakada ayrılmaya yol açtığı bir durumdur.
Prevalansı %1,65-72) olup, 40 yaş üstünde sık görülür ve yaklaşık %70’i bilateraldir. %70 oranında alt temporal periferik retinada yerleşir ve yüzeyi düzdür. Kalıtsal değildir, yaşlanmayla birlikte ortaya çıkar.
Sekonder retina şizisi, proliferatif membranlar, vitreus traksiyonu, kistik değişiklikler, intraretinal kanama, eksüdasyon ve inflamasyondan kaynaklanabilir. Spesifik nedenler arasında diyabetik retinopati, eski retina dekolmanı, yaşa bağlı makula dejenerasyonu, prematüre retinopatisi (ROP) ve Coats hastalığı yer alır.
Çoğu asemptomatiktir ve arka kutba ilerlemesi nadirdir; görme genellikle normaldir. Lezyon ekvatoru aştığında görme alanı defekti gibi semptomlar ortaya çıkabilir.
| Bulgular | Özellikler | Ayırıcı Tanıdaki Önemi |
|---|---|---|
| Kubbe şeklinde kabarıklık | Düzgün, saydam yarımküre; sabit ve hareketsiz | Pozisyon değişikliği ile hareket etmez |
| water silk appearance | İç tabakada dalgalı parlaklık | Bu hastalık için karakteristik |
| snowflakes (kar tanesi benzeri bulanıklıklar) | Ayrışma tabakasının iç yüzeyinde sarı-beyaz granüler bulanıklıklar | Dejenerasyon göstergesi |
Dış tabaka deliği oluşup retina dış tabaka dekolmanı gelişse bile risk düşüktür. İç ve dış delikler oluşup dekolman ilerlerse, tipik yırtıklı retina dekolmanı için cerrahi tedavi gerekir.
Ana patoloji, yaşlanmaya bağlı olarak dış pleksiform tabakadaki Henle liflerinin kistik dejenerasyonunun birleşmesi ve genişlemesidir. Mutlak skotomun mekanizması, dış pleksiform tabakadaki katman ayrışmasının sinaps bağlantılarında doku yırtılmasına yol açarak ışık uyarısının sinyal iletimini engellemesidir 1).
Risk faktörleri arasında yaşlanma ve hipermetropi yer alır 2). Genetik yatkınlık tanımlanmamıştır 2).
OCT ile tanı merkezi bir rol oynar2). Ayrılma boşluğunun yayılımı, derinliği ve katmanlar arasındaki sütun benzeri yapılar (dikey doku köprüleri) görüntülenir ve dış pleksiform tabakada katmanlar arası ayrılma doğrudan doğrulanabilir.
Retina dekolmanından ayırıcı tanı en önemlisidir.
| Özellik | Retinoskizis | Retina dekolmanı |
|---|---|---|
| Kabartının hareketliliği | Sabit, hareket etmez | Pozisyon değişikliği ile hareket eder |
| Geçirgenlik | Yüksek (saydam) | Düşük (bulanık) |
| Skotomun özelliği | Mutlak skotom | Rölatif skotom |
| Yüzey özellikleri | Düz (water silk) | Düzensiz, dalgalı |
Prensip olarak takip yeterlidir. Retina dekolmanına ilerleme riski yılda yaklaşık %0.05 gibi düşük olup, çoğu vaka asemptomatik ve stabildir.
Arkaya İlerleme Riski
Temel prensip: Gözlem
Bariyer lazer endikasyonu: İlerleyici genişleme, arka kutba yakınlık
Tedavi amacı: Daha fazla arka ilerlemenin önlenmesi
Dış tabaka yırtığı oluşumu
Temel prensip: Gözlem
Dikkat edilmesi gerekenler: Dış tabaka yırtığı dekolman riskini artırır
Endikasyonu dikkatlice değerlendirin: Aynı anda iç tabaka deliği varsa dikkatli olunmalıdır
Retinoskizis dekolmanı
Temel prensip: Genellikle yırtıklı retina dekolmanına benzer cerrahi
Cerrahi endikasyon: İç ve dış tabakalarda delik oluşması ve dekolmanın ilerlemesi durumunda
Prognoz: Genellikle iyi
Makula infiltrasyonu olgusu
Temel prensip: Vitrektomi cerrahisinin değerlendirilmesi
Cerrahi yöntem: ILM soyulması + gaz tamponadı
Kanıt düzeyi: Olgu sunumu düzeyi 1)
Dış pleksiform tabakadaki katmanlar arası ayrışma, sinaps bağlantılarında doku yırtılmasına yol açar ve mutlak skotom oluşur1). Henle liflerinin yaşa bağlı kistoid dejenerasyonun birleşmesiyle ayrılmasının, ayrışmanın nedeni olduğu düşünülmektedir. SD-OCT ile üç boyutlu değerlendirme, ayrışma boşluğunun yayılımını ve iç-dış tabakaların inceliğini hassas bir şekilde değerlendirmeyi mümkün kılmış, uzun vadeli doğal seyir ve ilerleme öngörücülerinin belirlenmesi ilerlemiştir2).
Yaşa bağlı retina ayrışmasında dış pleksiform tabakada katmanlar arası ayrışma meydana gelir. Bu bölgede fotoreseptörler ile ganglion hücreleri arasında sinaps bağlantıları bulunur ve ayrışma sonucu sinapslar koparsa, ışık uyarısı bipolar hücrelere ve ganglion hücrelerine iletilemez1). Bu nedenle, ayrışma alanında ışık tutulsa bile elektrik sinyali iletilmez ve mutlak skotom olarak hissedilir.
Çoğu yaşa bağlı retinoskizis stabil bir seyir izler. Retina dekolmanına ilerleme riski yılda yaklaşık %0.05 gibi son derece düşüktür. Ancak iç ve dış katmanlarda delik oluşursa dekolman riski artar, bu nedenle düzenli fundus muayenesi ile takip önemlidir2).
X’e bağlı retinoskizis (XLRS), RS1 gen mutasyonuna bağlı X kromozomu resesif geçişli konjenital bir retina hastalığıdır. Nispeten nadir bir vitreoretinal dejeneratif hastalık olup görülme sıklığı 5.000-25.000’de 1 olarak bildirilmiştir2). Genellikle sadece erkekler etkilenir, kadınlar taşıyıcıdır.
Genellikle sadece erkekler etkilenir. X kromozomu resesif geçişli olduğundan, kadınlarda sadece bir X kromozomunda mutasyon varsa taşıyıcı olurlar ve genellikle asemptomatiktirler. Nadiren, X kromozomu inaktivasyonunda sapma veya uniparental dizomi nedeniyle kadınlarda hafif fenotip görülebileceği bildirilmiştir2).
Belirtiler:
Klinik bulgular:
Makula bulguları
Foveal ayrılma (hemen hemen tüm vakalar): OCT’de kistler ve köprü yapıları ile birlikte tekerlek teli benzeri kıvrımlar gösteren kistoid değişiklikler
Makula atrofisi: Yaşla birlikte kist benzeri değişiklikler geriler ve atrofiye dönüşür; birçok olgu basit kistoid makula ödemi gibi görünür
Floresein anjiyografi: Kistoid makula ödeminden farklı olarak floresan sızıntısı yoktur (fovea normal veya pencere defekti)
Periferik Bulgular
Periferik retina ayrılması (yaklaşık yarısında): Alt temporal bölgede sık görülür; büyük iç retinal tabaka yırtıkları eşlik edebilir
Altın yaprak refleksi: Periferik retinada karakteristik bulgu; retina damarlarında beyazlaşma eşlik eder
Vitreus perdesi: Ağır vakalarda görülür; vitreus ayrılmamıştır ve retinaya sıkıca yapışıktır
Komplikasyonlar: Retina dekolmanı (%5-20), vitreus kanaması, ayrılma boşluğunda kanama
Erkek çocuklarda iki taraflı kistoid makula ödemi görüldüğünde bu hastalıktan şüphelenmek önemli bir tanı giriş noktasıdır.
Ayırıcı tanı:
| Hastalık | Ayırıcı tanı noktaları |
|---|---|
| Regmatojen retina dekolmanı | Genellikle tek taraflı; demarkasyon hattı oluşumu; lattice dejenerasyonu eşlik eder |
| Kistoid makula ödemi | Floresein anjiyografide petaloid sızıntı; XLRS’de sızıntı yok |
| Yaşa bağlı (dejeneratif) retina yırtılması | Yaşlılarda görülür; periferle sınırlı; b dalgası normal |
| Goldman-Favre sendromu | Gece körlüğü ve pigmentasyon eşlik eder |
Kistoid makula ödeminde (KMÖ) floresein anjiyografide petaloid (taç yaprağı şeklinde) floresan sızıntısı ve birikimi görülür. Buna karşılık XLRS’de foveal ayrışma boşluğu bulunur ancak kan-retina bariyeri normaldir; floresein anjiyografide sızıntı olmaz, sadece pencere defekti izlenir. Bu fark, OCT ve floresein anjiyografinin birlikte kullanıldığı ayırıcı tanıda önemlidir.
Kesinleşmiş bir küratif tedavi yoktur.
İlaç tedavisi:
Cerrahi tedavi:
Hasta yönetimi:
Büyümeyle birlikte foveadaki kist benzeri değişiklikler kendiliğinden azalabilir 2). Ancak makula atrofisine ilerleyen vakalar da vardır; gerileme her zaman görme iyileşmesi anlamına gelmez. Bazı vakalarda arka vitreus dekolmanı oluştuktan sonra periferik ayrılmanın azaldığı da bildirilmiştir.
RS1 geni tarafından kodlanan retinoskisin, 224 amino asitten oluşan bir salgı proteinidir ve diskoidin alanı aracılığıyla homo-oligomerik kompleksler oluşturur2). Fotoreseptör ve bipolar hücrelere bağlanarak retinanın yapısal ve sinaptik bütünlüğünü korur. RS1 mutasyonu kompleksin bozulmasına yol açtığında karakteristik ayrışma boşlukları ve retina sinyal iletim bozukluğu ortaya çıkar2).
Histopatolojik olarak retina ayrışması esas olarak sinir lifi tabakasında meydana gelir. OCT’de fovea ve çevresinde dış pleksiform tabaka ile iç granüler tabakada ayrışma görülür. Morfolojik ayrışma sınırlı olsa bile, elektrofizyolojik olarak tüm retinada fonksiyon bozukluğu olduğu negatif b dalgasından anlaşılır.
Genotip-fenotip korelasyonu (83 Koreli hasta kohortu, Lee ve ark. 2025)2):
XLRS ayrışma sıvısını analiz eden çalışmalarda, fotoreseptörler arası retinoid bağlayıcı protein (IRBP) ayrışma sıvısında tanımlanmış olup, ayrışma boşluğunda retinal metabolizma anormalliğine işaret edebileceği bildirilmiştir2). Ayrıca XLRS’de genellikle görülmeyen eksüdatif makülopatinin eşlik ettiği vakalar da rapor edilmiş olup, aynı tanı altında çeşitli fenotiplerin varlığı gösterilmiştir2).
RS1 gen tedavisi için klinik çalışmalar devam etmektedir2)3). Hayvan modellerinde ve erken klinik çalışmalarda elektroretinogram yanıtlarında kısmi iyileşme ve retina anatomisinde düzelme bildirilmiştir.
Başlıca klinik çalışmalar:
Mevcut zorluk: XLRS’de retina kırılganlığı artmıştır ve arka vitreus membranının ayrılması ile subretinal enjeksiyon teknik olarak çok zordur3). İntravitreal uygulama bazen tercih edilebilir, ancak vitritis riski kabul edilmelidir.
XLRS için gen tedavisi henüz klinik deneme aşamasındadır ve genel hastanelerde tedavi olarak onaylanmamıştır. Bazı klinik çalışmalar göz içi inflamasyonu gibi güvenlik sorunlarını ortaya çıkarmış ve bazı denemeler durdurulmuştur3). Katılmak isteyenlerin uzman bir merkeze danışması gerekir.
Miyopik foveoskizis, yüksek miyopiye bağlı olarak arka kutupta retina iç tabakalarının ayrılmasıdır. Eskiden yüksek miyopiye eşlik eden arka kutup retina dekolmanı olarak tanımlanmış olup, günümüzde makula deliği ile birlikte olanları da içeren bir kavram haline gelmiştir.
Patofizyolojide aşağıdaki faktörler karmaşık bir şekilde rol oynar:
Semptomlar: Görme keskinliğinde azalma, santral skotom, metamorfopsi, bulanık görme. Yüksek miyopide semptomlar hafif olabilir.
Tanı (OCT):
Tedavi: Vitrektomi temel tedavidir.
Prognoz: