Bệnh vô mống mắt (aniridia) là tình trạng mống mắt bị thiếu hụt hoàn toàn hoặc không hoàn toàn do yếu tố bẩm sinh. Mặc dù được gọi là ‘vô mống mắt’, nhưng thường vẫn còn sót lại phần gốc mống mắt ở vùng ngoại vi nhất của góc tiền phòng.
Năm 2017, bệnh được công nhận là bệnh hiếm được chỉ định theo Luật Bệnh hiếm của Bộ Y tế, Lao động và Phúc lợi Nhật Bản 1). Bệnh nhân được chẩn đoán mắc bệnh hiếm được chỉ định và được đánh giá mức độ nghiêm trọng từ độ III trở lên sẽ được hỗ trợ chi phí y tế, với mức trần tự chi trả dựa trên thu nhập 2).
Khoảng 2/3 tổng số (di truyền trội trên nhiễm sắc thể thường)
Lẻ tẻ
Khoảng 1/3 tổng số
Kết hợp u Wilms (trường hợp lẻ tẻ)
Khoảng 30% (hội chứng WAGR)3)
Các nghiên cứu dịch tễ học tại Thụy Điển và Na Uy báo cáo tỷ lệ mắc bệnh khoảng 1 trên 90.000 người3). Đánh giá nhãn khoa chi tiết trên 43 bệnh nhân có đột biến gen PAX6 cho thấy mức độ bất thường của mống mắt khác nhau tùy theo loại đột biến3).
QBệnh vô mống mắt có di truyền không?
A
Khoảng 2/3 tổng số trường hợp là di truyền trội trên nhiễm sắc thể thường, với 50% khả năng truyền từ cha mẹ bị bệnh sang con. 1/3 còn lại là lẻ tẻ, không có tiền sử gia đình. Ở các trường hợp lẻ tẻ, có nguy cơ mắc hội chứng WAGR kèm u Wilms (u thận), do đó khuyến nghị xét nghiệm di truyền gen PAX6 và WT1.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
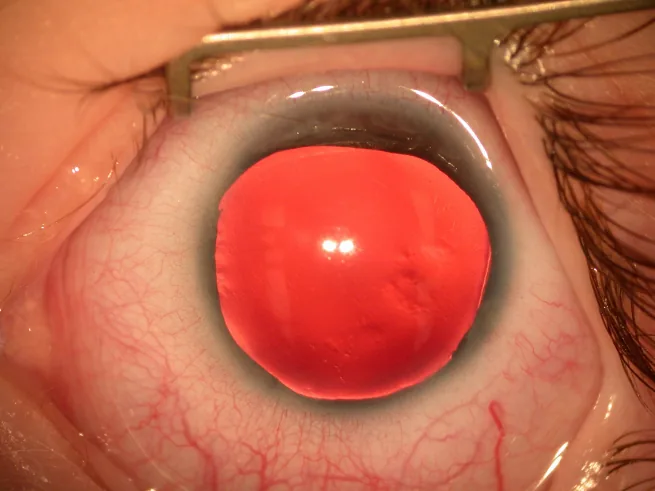
Ảnh chụp đèn khe phần trước nhãn cầu cho thấy mống mắt gần như khuyết hoàn toàn, chỉ còn một ít mống mắt rất mỏng ở ngoại vi. Hình ảnh này trực tiếp cho thấy dấu hiệu lâm sàng điển hình của bệnh vô mống mắt, phù hợp để mô tả các triệu chứng chính và dấu hiệu lâm sàng.
Do mống mắt bị khuyết hoặc không hoàn chỉnh, đồng tử không hoạt động, không thể điều chỉnh lượng ánh sáng vào mắt. Vì vậy, bệnh nhân thường phàn nàn về chứng sợ ánh sáng mạnh. Ngoài ra, tình trạng cố định thị lực kém do bất sản hoàng điểm thường là nguyên nhân chính gây rung giật nhãn cầu ngang xuất hiện sớm sau sinh.
Sợ ánh sáng: Không thể điều chỉnh lượng ánh sáng qua mống mắt → chói mắt mạnh
Gen PAX6 biểu hiện không chỉ ở mô mắt mà còn ở hệ thần kinh trung ương, đảo tụy Langerhans và biểu mô khứu giác. Sự giảm sản của các mô này có thể dẫn đến nhiều biến chứng ngoài mắt khác nhau 1).
Thiểu sản thể chai, động kinh, rối loạn chức năng não bậc cao
Mất khứu giác
Không dung nạp glucose
Hội chứng WAGR (khoảng 30% ca lẻ tẻ): U Wilms, vô mống mắt, bất thường cơ quan sinh dục niệu, chậm phát triển tâm thần3)
QBệnh vô mống mắt nhìn được bao nhiêu?
A
Tiên lượng thị lực nhìn chung kém, thường khoảng 0,1. Tuy nhiên, tùy thuộc vào mức độ giảm sản hoàng điểm và sự hiện diện của biến chứng, thị lực có thể dao động từ 0,1 đến 0,7. Giảm sản hoàng điểm hiện chưa có phương pháp điều trị hiệu quả và là yếu tố hạn chế thị lực lớn nhất. Chỉnh khúc xạ phù hợp và chăm sóc thị lực kém có thể cải thiện chất lượng cuộc sống hàng ngày.
Nguyên nhân của vô mống mắt là do mất chức năng một alen (bất hoạt một alen) của gen PAX6 nằm trên nhánh ngắn nhiễm sắc thể số 11 (11p13). Tình trạng này xảy ra do lượng gen chức năng giảm một nửa. Nếu cả hai alen đều bất thường, được cho là gây chết phôi thai1).
PAX6 là gen kiểm soát chính của yếu tố phiên mã điều khiển sự biệt hóa cơ quan trong giai đoạn phôi thai, chi phối nhiều yếu tố phiên mã khác nhau. Bất thường của PAX6 gây ra nhiều dị tật bẩm sinh trên toàn bộ nhãn cầu (vô mống mắt, bất thường Peters, giảm sản hoàng điểm, v.v.).
Các dạng đột biến gen thường là loại mã kết thúc sớm (PTC) như vô nghĩa, dịch khung; đột biến sai nghĩa cũng đã được báo cáo1). Phân tích giải trình tự ở vô mống mắt đơn độc phát hiện đột biến PAX6 ở khoảng 85% trường hợp2).
Gen PAX6 nằm liền kề với gen ức chế khối u WT1 trên nhiễm sắc thể 11p13. Ở các ca lẻ tẻ, mất đoạn gen liền kề có thể gây ra hội chứng WAGR bao gồm u Wilms, vô mống mắt, bất thường cơ quan sinh dục niệu và chậm phát triển tâm thần3). Khoảng 30% ca lẻ tẻ có u Wilms xuất hiện sớm hai bên trước 5 tuổi.
Dương tính với đột biến PAX6, không mất WT1 → có thể ước tính không có khả năng mắc hội chứng WAGR2)
Xét nghiệm di truyền kết hợp giải trình tự DNA và phát hiện bất thường cấu trúc gen bằng MLPA/CMA2)
Đối với trường hợp lẻ tẻ nghi ngờ hội chứng WAGR, xét nghiệm di truyền được khuyến nghị 2)
QCó nên thực hiện xét nghiệm di truyền cho bệnh vô mống mắt không?
A
Xét nghiệm gen PAX6 cần thiết để xác định chẩn đoán Definite, đặc biệt ở các trường hợp lẻ tẻ, xét nghiệm di truyền PAX6 và WT1 được khuyến nghị để đánh giá nguy cơ u Wilms. Việc xét nghiệm nên kết hợp giải trình tự DNA với MLPA/CMA và được thực hiện dưới sự tư vấn di truyền phù hợp.
Tầm soát u Wilms (trường hợp lẻ tẻ, vài tháng một lần, đặc biệt đến 5 tuổi)
Xét nghiệm di truyền
Xác định đột biến gen PAX6 hoặc mất đoạn vùng 11p13 (cần thiết cho chẩn đoán xác định)
Ở trẻ em, có thể cần kiểm tra dưới gây mê toàn thân.
QChẩn đoán bệnh vô mống mắt được thực hiện như thế nào?
A
Cơ bản là xác nhận bất thường mống mắt bằng khám đèn khe và đánh giá giảm sản hoàng điểm bằng OCT. Xét nghiệm gen PAX6 có thể chẩn đoán xác định, và ở trường hợp lẻ tẻ cần kết hợp tìm kiếm gen WT1. Cần phân biệt với teo mống mắt do herpes, khuyết mống mắt sau chấn thương, u mống mắt, bất thường Rieger và hội chứng ICE.
Bất thường mống mắt, giảm sản hoàng điểm, mắt nhỏ và rung giật nhãn cầu hiện không thể can thiệp, chủ yếu theo dõi. Đối tượng điều trị là bệnh giác mạc, đục thủy tinh thể, glôcôm, sợ ánh sáng và thị lực kém2).
Đục giác mạc nhu mô: Cải thiện chức năng thị giác nhờ ghép giác mạc bị hạn chế do các biến chứng của bệnh vô mống mắt2). Về lâu dài, tiên lượng thị lực thường xấu do bệnh tăng nhãn áp nặng hơn và suy chức năng mảnh ghép theo thời gian. Ghép giác mạc xuyên toàn bộ cho đục giác mạc thường không cải thiện thị lực và cần lưu ý tỷ lệ thải ghép cao. Ở các trường hợp nặng, cần cân nhắc kỹ lợi ích và tác hại trước khi quyết định thực hiện.
Suy kiệt tế bào gốc biểu mô giác mạc (LSCD): Cân nhắc điều trị phẫu thuật 2). Cụ thể, ghép rìa giác mạc đồng loại (KLAL) hoặc ghép biểu mô niêm mạc miệng nuôi cấy (COMET) có thể giúp tái tạo bề mặt nhãn cầu ở một mức độ nhất định 3). Khi có kèm đục giác mạc nhu mô, kết hợp ghép giác mạc thường hữu ích để cải thiện thị lực2).
Tăng nhãn áp ảnh hưởng trực tiếp đến tiên lượng thị lực, do đó cần điều trị tích cực 2). Áp dụng các phương pháp tiếp cận theo từng bước sau:
Điều trị nội khoa: Hạ nhãn áp bằng thuốc nhỏ mắt hoặc uống, lưu ý tác dụng phụ và ảnh hưởng toàn thân ở trẻ em
Phẫu thuật tái tạo đường dẫn lưu: Mở góc tiền phòng hoặc cắt bè (cân nhắc khi điều trị nội khoa không hiệu quả)
Phẫu thuật lọc: Cắt bè củng giác mạc
Phẫu thuật cấy ghép glôcôm: Phẫu thuật ống dài (cần chứng nhận cơ sở)
Phẫu thuật đông lạnh thể mi: Biện pháp cuối cùng khi các phương pháp khác không hiệu quả
Kháng thuốc thường gặp, và phẫu thuật dẫn lưu ống có thể là lựa chọn tốt4). Vì tổn thương thị trường do glôcôm không hồi phục, kiểm soát nhãn áp sớm là chìa khóa bảo tồn chức năng thị giác.
QGlôcôm do vô mống mắt được điều trị như thế nào?
A
Đầu tiên, điều trị bằng thuốc nhỏ mắt hoặc uống, nhưng thường kháng thuốc. Nếu không hiệu quả, cân nhắc phẫu thuật tái tạo đường lưu thông (phẫu thuật mở góc tiền phòng, phẫu thuật mở bè củng mạc), sau đó tiến tới phẫu thuật cắt bè củng mạc hoặc phẫu thuật ống dài (cấy ghép glôcôm). Phẫu thuật ống dài cần chứng nhận cơ sở. Phẫu thuật đông lạnh thể mi là biện pháp cuối cùng khi các phương pháp khác không hiệu quả. Theo dõi nhãn áp định kỳ là cần thiết.
Gen PAX6 là gen kiểm soát chính mã hóa yếu tố phiên mã điều phối sự biệt hóa cơ quan trong giai đoạn phôi thai. Nó được biểu hiện từ giai đoạn đầu của mắt và điều phối nhiều yếu tố phiên mã khác. Mất chức năng một alen của PAX6 (haploinsufficiency) gây ra các dị tật bẩm sinh toàn bộ nhãn cầu (như vô mống mắt, dị tật Peters, giảm sản hoàng điểm).
Đột biến PAX6 thường là loại PTC như vô nghĩa hoặc dịch khung, và đột biến sai nghĩa cũng đã được báo cáo 1). Các nghiên cứu về tương quan kiểu gen - kiểu hình cho thấy mức độ nghiêm trọng của các biểu hiện nhãn khoa khác nhau tùy theo loại đột biến 3).
PAX6 cũng được biểu hiện ở hệ thần kinh trung ương, đảo tụy Langerhans và biểu mô khứu giác, và có thể gây ra các biến chứng ngoài mắt (thiểu sản thể chai, động kinh, mất khứu giác, không dung nạp glucose) do giảm sản các mô này 1).
Bệnh lý góc mở: Tăng sức cản dòng chảy thủy dịch ở bè củng mạc
Bệnh lý góc đóng: Phần gốc mống mắt còn sót lại ở vùng ngoại vi nhất dính vào bè củng mạc, tạo ra một dạng bệnh lý glôcôm góc đóng
Glôcôm hiếm khi xuất hiện ở giai đoạn sơ sinh, thường tiến triển dần ở tuổi thanh niên. Nó có thể xảy ra do bất thường góc tiền phòng gây góc mở hoặc do góc đóng.
Về mặt bệnh lý, có rối loạn chức năng tế bào gốc biểu mô giác mạc, gây bất thường biểu mô và màng Bowman, hình thành màng máu (pannus) giàu mạch. Sự kém phát triển của các palisades Vogt dẫn đến xâm lấn mô kết mạc và hóa sừng 1).
Giác mạc ở bệnh nhân vô mống mắt dày hơn so với người bình thường. Ở trẻ nhỏ, giác mạc thường bình thường, nhưng khi lớn lên, có thể xuất hiện đục nhu mô giác mạc và LSCD, gây giảm thị lực. Trong một nghiên cứu đơn trung tâm kéo dài 14 năm (738 mắt), vô mống mắt chiếm 30,9% các nguyên nhân gây LSCD, cao nhất 6).
Tiên lượng thị lực nhìn chung kém, thường khoảng 0,1
Giảm sản hoàng điểm không có phương pháp điều trị hiệu quả và là yếu tố hạn chế thị lực lớn nhất
Tổn thương thị trường do glôcôm là không hồi phục, do đó kiểm soát nhãn áp sớm rất quan trọng
Đối với các trường hợp lẻ tẻ, cần chú ý đến sự khởi phát sớm của u Wilms trước 5 tuổi và tiếp tục siêu âm bụng định kỳ.
Các nghiên cứu về tiên lượng dài hạn báo cáo rằng tiên lượng thị lực nhìn chung là kém, nhưng có sự khác biệt cá nhân tùy thuộc vào loại và mức độ nghiêm trọng của biến chứng5).
Với sự phổ biến của giải trình tự thế hệ mới (NGS), tỷ lệ phát hiện đột biến PAX6 trong aniridi đơn độc đạt khoảng 85%2). Microarray nhiễm sắc thể (CMA) có độ nhạy cao hơn so với xét nghiệm nhiễm sắc thể truyền thống trong việc phát hiện mất đoạn nhỏ 11p13, góp phần cải thiện độ chính xác chẩn đoán hội chứng WAGR2).
Kết quả dài hạn của ghép biểu mô niêm mạc miệng nuôi cấy (COMET) đang được tích lũy2). Đối với Boston type I keratoprosthesis, cải thiện thị lực đạt được 65–93% trong ngắn hạn (17–28,7 tháng), nhưng giảm xuống 43,5% tại thời điểm 4,5 năm theo một báo cáo2).
Thiết bị mống mắt nhân tạo và triển vọng liệu pháp gen
HumanOptics CustomFlex ArtificialIris là thiết bị mống mắt nhân tạo bằng silicon được đặt hàng riêng, hữu ích trong việc giảm chói và cải thiện thẩm mỹ, nhưng chưa được phê duyệt tại Nhật Bản tính đến năm 2024. Liệu pháp nhắm mục tiêu phân tử nhắm vào tình trạng thiếu hụt PAX6 hiện đang trong giai đoạn nghiên cứu và chưa được ứng dụng lâm sàng3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.