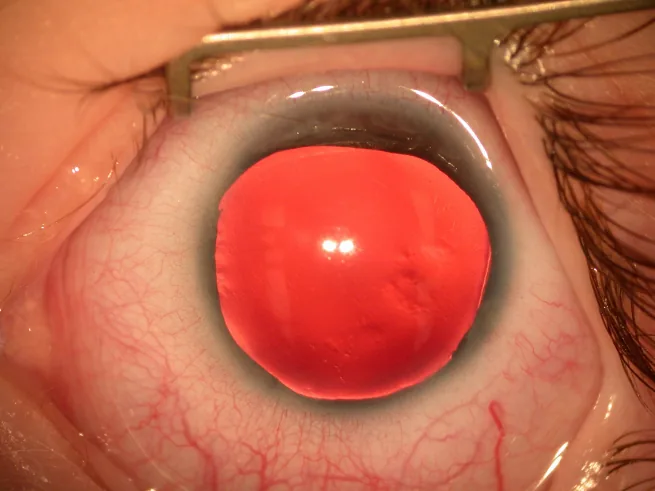
Aniridia is a condition in which the iris is completely or partially absent due to a congenital predisposition. Although called “aniridia,” a remnant of the iris root often remains at the most peripheral part of the angle.
In 2017, it was designated as an intractable disease under the Ministry of Health, Labour and Welfare’s Intractable Disease Act 1). Patients diagnosed with the designated intractable disease and judged to have a severity classification of grade III or higher are eligible for medical expense subsidies, with a maximum out-of-pocket amount set according to income 2).
Epidemiological studies in Sweden and Norway report a prevalence of approximately 1 in 90,000 people 3). Detailed ophthalmologic evaluation of 43 patients with PAX6 gene mutations showed that the degree of iris dysplasia varies depending on the type of mutation 3).
QIs aniridia inherited?
A
About two-thirds of cases are autosomal dominant, with a 50% chance of inheritance from an affected parent to a child. The remaining one-third are sporadic with no family history. In sporadic cases, there is a risk of WAGR syndrome, which includes Wilms tumor (kidney tumor), so genetic testing of the PAX6 and WT1 genes is recommended.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Slit-lamp photograph of the anterior segment showing the iris almost completely absent, with only a very thin remnant of iris visible at the periphery. This directly shows the typical clinical findings of aniridia and is suitable for explaining the main symptoms and clinical findings.
Because the iris is absent or incomplete, the pupil does not function, and the amount of light entering the eye cannot be regulated. Therefore, patients complain of severe photophobia. Additionally, poor fixation due to foveal hypoplasia often presents as horizontal nystagmus from early infancy.
Photophobia: Inability to regulate light due to iris defect → severe glare
The PAX6 gene is expressed not only in ocular tissues but also in the central nervous system, pancreatic islets of Langerhans, and olfactory epithelium. Hypoplasia of these tissues can lead to various extraocular complications 1).
Agenesis of the corpus callosum, epilepsy, higher brain dysfunction
Anosmia
Glucose intolerance
WAGR syndrome (approximately 30% of sporadic cases): Wilms tumor, aniridia, genitourinary abnormalities, and intellectual disability3)
QHow well can people see with aniridia?
A
Visual prognosis is generally poor, often around 0.1. However, depending on the degree of foveal hypoplasia and the presence of complications, visual acuity can range from 0.1 to 0.7. Currently, there is no effective treatment for foveal hypoplasia, which is the main limiting factor for vision. Appropriate refractive correction and low vision care can improve quality of daily life.
Aniridia is caused by loss of function (haploinsufficiency) of one allele of the PAX6 gene located on the short arm of chromosome 11 (11p13). It results from a reduction in functional gene dosage. Biallelic abnormalities are thought to be embryonic lethal1).
PAX6 is a master control gene encoding a transcription factor that governs organ differentiation during embryonic development and regulates various other transcription factors. Abnormalities in PAX6 lead to various congenital anomalies throughout the eye, including aniridia, Peters anomaly, and foveal hypoplasia.
The types of genetic mutations are often premature truncated codon (PTC) mutations such as nonsense and frameshift mutations, and missense mutations have also been reported1). Sequencing analysis of isolated aniridia detects PAX6 mutations in approximately 85% of cases2).
WAGR syndrome (important considerations in sporadic cases)
The PAX6 gene is adjacent to the tumor suppressor gene WT1 on chromosome 11p13. In sporadic cases, contiguous gene deletions can cause WAGR syndrome, which consists of Wilms tumor, aniridia, genitourinary abnormalities, and intellectual disability3). Approximately 30% of sporadic cases develop early bilateral Wilms tumor by age 5.
PAX6 mutation positive and no WT1 deletion → WAGR syndrome is unlikely2)
Genetic testing should combine DNA sequencing with MLPA/CMA to detect genomic structural abnormalities2)
Genetic testing is recommended for sporadic cases suspected of WAGR syndrome 2)
QShould I undergo genetic testing for aniridia?
A
PAX6 genetic testing is necessary to confirm a definite diagnosis, and especially in sporadic cases, genetic testing for PAX6 and WT1 is recommended to assess Wilms tumor risk. It is important to perform testing using a combination of DNA sequencing and MLPA/CMA, under appropriate genetic counseling.
Wilms tumor screening (sporadic cases, every few months, especially until age 5)
Genetic testing
Identification of PAX6 gene mutation or 11p13 deletion (required for definite diagnosis)
In children, examination under general anesthesia may be necessary.
QHow is aniridia diagnosed?
A
The basics include confirming iris dysplasia with slit-lamp microscopy and evaluating foveal hypoplasia with OCT. Definite diagnosis is possible with PAX6 genetic testing, and in sporadic cases, WT1 gene analysis is also performed. Differentiation from herpes iris atrophy, traumatic iris defect, iris coloboma, Rieger anomaly, and ICE syndrome is important.
Corneal stromal opacity: Visual improvement achieved by corneal transplantation is limited due to complications of aniridia2). Long-term visual prognosis is often poor due to worsening glaucoma and progressive graft failure over time. Full-thickness corneal transplantation for corneal opacity often does not lead to visual improvement, and attention should be paid to the high rate of rejection. In severe cases, the decision to perform surgery should be made after carefully considering the balance of benefits and harms.
Limbal stem cell deficiency (LSCD): Surgical treatment should be considered 2). Specifically, keratolimbal allograft (KLAL) or cultivated oral mucosal epithelial transplantation (COMET) can provide some degree of ocular surface reconstruction3). When corneal stromal opacity is also present, combining corneal transplantation is often useful for visual improvement 2).
Glaucoma is directly linked to visual prognosis, so aggressive treatment is warranted 2). A stepwise approach is taken as follows.
Medical therapy: Lower intraocular pressure using eye drops and oral medications, paying attention to side effects and considering systemic effects in children
Outflow reconstruction surgery: Goniotomy or trabeculotomy (considered when medical therapy is ineffective)
Glaucoma implant surgery: Long tube surgery (facility certification required)
Cyclophotocoagulation: Last resort when other treatments fail
There is often resistance to drug therapy, and tube shunt surgery may be a good option4). Since glaucoma causes irreversible visual field damage, early intraocular pressure management is key to preserving visual function.
QHow is glaucoma in aniridia treated?
A
First, drug therapy with eye drops or oral medications is performed, but many cases are resistant to drugs. If the effect is insufficient, outflow reconstruction surgery (goniotomy or trabeculotomy) is considered, followed by trabeculectomy or long tube surgery (glaucoma implant surgery). Long tube surgery requires facility certification. Cyclophotocoagulation is the last resort when other treatments fail. Regular intraocular pressure monitoring is essential.
The PAX6 gene is a master control gene that encodes a transcription factor governing organ differentiation during the embryonic period. It is expressed from the early eye and regulates various transcription factors. Loss of function of one allele of PAX6 (haploinsufficiency) causes congenital abnormalities throughout the eye (aniridia, Peters anomaly, macular hypoplasia, etc.).
PAX6 mutations are often of the PTC type, such as nonsense and frameshift mutations, and missense mutations have also been reported 1). Studies on genotype-phenotype correlation have shown that the severity of ophthalmic findings varies depending on the type of mutation 3).
PAX6 is also expressed in the central nervous system, pancreatic islets of Langerhans, and olfactory epithelium, and hypoplasia of these tissues can lead to extraocular complications (corpus callosum agenesis, epilepsy, anosmia, glucose intolerance) 1).
Open-angle pathology: Increased resistance to aqueous humor outflow in the trabecular meshwork
Angle-closure pathology: The residual iris root at the periphery adheres to the trabecular meshwork, leading to a type of angle-closure glaucoma
Glaucoma rarely presents in infancy and progressively develops in adolescence with growth. It may occur in an open state due to angle dysgenesis or present as glaucoma due to angle closure.
Pathologically, functional abnormalities of corneal epithelial stem cells are observed, leading to abnormalities in the epithelium and Bowman’s membrane, and formation of a vascularized pannus. Hypoplasia of the palisades of Vogt progresses to conjunctival tissue invasion and keratinization 1).
The cornea in aniridia is thicker than in healthy individuals. The cornea is often normal in early childhood, but with growth, corneal stromal opacity and LSCD develop, causing visual impairment. In a 14-year single-center study (738 eyes), aniridia was the most common cause of LSCD, accounting for 30.9% of cases 6).
Visual prognosis is generally poor, often around 0.1
Macular hypoplasia has no effective treatment and is the greatest limiting factor for vision
Visual field damage due to glaucoma is irreversible, and early intraocular pressure management is important
In sporadic cases, be alert for early onset of Wilms tumor before age 5 and continue regular abdominal ultrasound examinations.
Studies on long-term prognosis report that visual prognosis is generally poor, but varies among individuals depending on the type and severity of complications5).
With the widespread use of next-generation sequencing (NGS), the detection rate of PAX6 mutations in isolated aniridia is approximately 85%2). Chromosomal microarray (CMA) is more sensitive than conventional chromosome testing for detecting microdeletions in 11p13, contributing to improved diagnostic accuracy for WAGR syndrome2).
Long-term outcomes of cultivated oral mucosal epithelial transplantation (COMET) are accumulating2). For the Boston type I keratoprosthesis, visual improvement is achieved in 65–93% of cases in the short term (17–28.7 months), but decreases to 43.5% at 4.5 years2).
Artificial Iris Devices and Gene Therapy Prospects
The HumanOptics CustomFlex ArtificialIris is a custom-made silicone artificial iris device that is useful for reducing photophobia and improving appearance, but as of 2024, it is not approved in Japan. Molecular targeted therapy for PAX6 haploinsufficiency is currently in the research stage and has not yet reached clinical application3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.