ICE syndrome (iridocorneal endothelial syndrome) is a disease in which abnormal corneal endothelial cells extend into the angle and iris with membranous tissue, obstructing the trabecular meshwork and causing increased intraocular pressure. Corneal endothelial abnormalities cause corneal edema, and contraction of the membranous tissue leads to iris abnormalities and peripheral anterior synechiae (PAS).
It occurs unilaterally, predominantly in women in their 30s to 40s 2). It is not hereditary and is sporadic. Family history is rare, and there is no consistent association with other ocular or systemic diseases.
ICE syndrome is classified into the following three clinical types based on the state of iris abnormalities.
Progressive iris atrophy: Characterized by marked iris atrophy, pupillary distortion, ectropion uveae, and multiple iris hole formation. This type most frequently accompanies glaucoma.
Chandler syndrome: Most common (about 50%). Corneal edema due to corneal endothelial dysfunction is the main feature, while pupillary deviation and iris atrophy are mild. Corneal edema tends to occur even with mild intraocular pressure elevation.
Cogan-Reese syndrome (iris nevus syndrome): Characterized by pigmented nodules on the anterior iris surface, with two types: pedunculated nodules and flat pigmented lesions 4). It is the rarest subtype.
QHow are the three types of ICE syndrome differentiated?
A
The differentiation of the three types is mainly based on clinical findings of the iris and cornea. Progressive iris atrophy is characterized by polycoria, pupillary deviation, iris hole formation, and ectropion uveae, with the highest rate of glaucoma complications. Chandler syndrome primarily presents with corneal edema and minimal iris changes, no iris holes, and low PAS. Cogan-Reese syndrome is characterized by pedunculated nodules or flat pigmented lesions on the anterior iris surface, and iris atrophy is usually absent. The basic pathophysiology is common to all types, and the treatment strategy is similar.
Unilateral visual loss (blurred vision due to corneal edema) is the most common initial symptom. Photophobia and pain may be present. Some patients visit the clinic after noticing external abnormalities such as pupillary deviation or iris heterochromia. Pain due to corneal edema and pain due to intraocular pressure elevation from angle closure may coexist.
Clinical Findings (Findings Confirmed by Physician Examination)
Corneal findings: In Chandler syndrome, the corneal endothelium exhibits a metallic sheen known as “hammered silver appearance.” It resembles the corneal guttata of Fuchs endothelial corneal dystrophy, but can be differentiated by its unilaterality. Corneal edema may occur even when intraocular pressure is within the normal range. Folds in Descemet’s membrane may also be observed.
Iris findings: Depending on the type, the following findings appear2):
Progressive Iris Atrophy
Pupillary deviation: The pupil is pulled toward the direction of extensive PAS.
Iris hole formation: Multiple holes develop in the direction opposite to the pupillary traction.
Uveal ectropion: The iris pigment epithelium becomes exposed on the surface.
PAS: Tall PAS extending beyond Schwalbe’s line are characteristic.
Glaucoma complication: Most frequent among the three types.
Chandler syndrome
Corneal edema: Easily occurs even with mild elevation of intraocular pressure; most prominent among the three types.
Iris findings: Pupillary deviation and iris atrophy are mild. No iris holes are observed.
Angle findings: Gonioscopy reveals PAS extending high beyond the Schwalbe line, a characteristic finding of ICE syndrome5). PAS are patchy and located very anteriorly, with the trabecular meshwork between PAS appearing normal5). The shape of PAS varies, including tent-shaped, trapezoidal, and broad adhesions.
The true etiology of ICE syndrome remains unknown, but there is a hypothesis that latent infection with herpes simplex virus (HSV) induces low-grade inflammation at the corneal endothelial level, leading to epithelial-like activation. PCR testing has reported detection of HSV DNA in over 60% of corneal and aqueous humor samples from ICE patients 2). Involvement of Epstein-Barr virus (EBV) has also been suggested.
Pathologically, normal endothelial cells are replaced by epithelial-like cells with migratory properties. Electron microscopy reveals epithelial features such as desmosomes, tonofilaments, and microvilli. Toxic damage (necrotic changes) to adjacent normal endothelial cells has also been reported.
Histopathological examination of Descemet’s membrane excised from ICE syndrome eyes reveals a monolayer of low cuboidal cells with partial bilayering or multilayering, and immunohistochemistry shows a mixed expression pattern of epithelial markers (AE1/AE3, CK8/18) and endothelial markers (CD56, vimentin) 7). This finding provides pathological evidence for the epithelial-like metaplasia that is the essence of ICE syndrome.
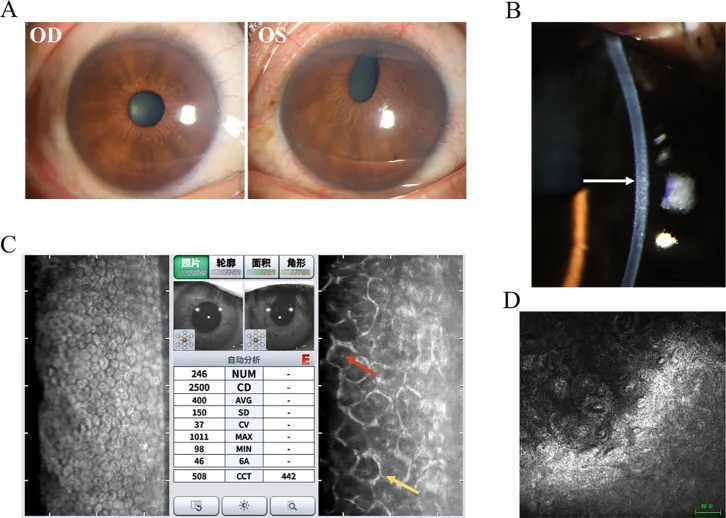
Hua Ma; Mingfang Xia; Qing Gu; et al. Iridocorneal endothelial syndrome. Front Ophthalmol (Lausanne). 2025;5:1655669. Figure 1. PMCID: PMC12537377. License: CC BY.
This figure summarizes the representative findings of Chandler syndrome. Slit-lamp photographs are presented alongside ancillary test results, making it easy to grasp what to confirm in ICE syndrome.
This is the most important test for confirming the diagnosis of ICE syndrome 2). Using a specular microscope, abnormal cells (ICE cells) with dark areas within the cell are observed for diagnosis. The normal hexagonal endothelial mosaic is lost, showing pleomorphism and a “light-dark reversal” pattern. Characteristically, the endothelial cells are larger and darker than normal, with a bright central spot.
A marked decrease in corneal endothelial cell density is observed. Normal values are approximately 3,500 cells/mm² in young individuals and 2,500–3,000 cells/mm² in the elderly, but when it drops below 500 cells/mm², pump function fails, leading to bullous keratopathy. In one case of Cogan-Reese syndrome, a decrease to 763 cells/mm² has been reported3).
In Chandler syndrome, specular microscopy reveals extensive pleomorphism and polymorphism of cells, and the borders of endothelial cells are indistinct, which aids in diagnosis. A coefficient of variation (CV) of 0.35 or higher is considered abnormal, and a hexagonality rate of 50% or less is considered abnormal.
It reveals cobblestone-like swollen endothelial cells, loss of hexagonal structure, highly reflective pleomorphic cells, and mononuclear or binuclear giant endothelial cells2). It is also useful when specular microscopy is difficult due to corneal edema.
It visualizes iridocorneal angle adhesions, hyperreflective thickening of the corneal endothelial layer, and membranous tissue on the iris2). It is excellent for quantitative evaluation of the extent and height of PAS.
It is essential for evaluating PAS and is used for diagnosis and follow-up of secondary angle-closure glaucoma. In ICE syndrome, PAS extending beyond Schwalbe’s line is characteristic, and the trabecular meshwork between PAS appears normal, which is useful for differentiation from POAG5).
Intraocular pressure measurement, optic disc photography, visual field testing (Humphrey or Goldmann), and OCT evaluation of retinal nerve fiber layer (RNFL) thickness are introduced for initial detailed examination and follow-up 2).
Differentiation from Cogan-Reese syndrome is necessary
QWhy is ICE syndrome sometimes misdiagnosed as open-angle glaucoma?
A
The advancing corneal endothelial membrane may functionally close the trabecular meshwork without causing contraction. In such cases, gonioscopy does not reveal obvious PAS, leading to a misdiagnosis of open-angle glaucoma despite the presence of “functional” angle closure. When encountering unilateral glaucoma, ICE syndrome should be included in the differential diagnosis, and the corneal endothelium should be carefully evaluated using specular microscopy.
Treatment of ICE syndrome consists of two main components: controlling intraocular pressure (IOP) in glaucoma and managing corneal decompensation. Long-term IOP control with medication is often difficult, and many cases ultimately require surgical intervention.
Medical Treatment
Aqueous humor suppressants: Topical beta-blockers, alpha-agonists, and carbonic anhydrase inhibitors (CAIs) are first-line treatments 6). Prescribe according to the regimen for primary open-angle glaucoma (POAG). Miotics targeting aqueous outflow are not recommended due to poor efficacy.
Prostaglandin analogs: Based on the hypothesis of latent HSV infection, there is concern about latanoprost-induced HSV reactivation, so use with caution 2).
Corneal edema management: Use hypertonic saline eye drops or gel to dehydrate the cornea.
Long-term prognosis: As the membranous tissue progresses and PAS expands, drug treatment often becomes resistant 6).
Surgical Treatment
Trabeculectomy: Performed with antifibrotic agents (mitomycin C; MMC or 5-FU) 6). Survival rates are reported as 73% at 1 year, 44% at 3 years, and 29% at 5 years. There is a risk of fistula occlusion by the abnormal endothelial membrane, and the success rate is low.
Tube shunt surgery: Tube shunt surgery with a plate is often chosen 5)6). Survival rates are 71% at 1 year, 71% at 3 years, and 53% at 5 years, showing better long-term outcomes than trabeculectomy.
Cyclodestructive procedures: Semiconductor laser cyclophotocoagulation (diode CPC) is considered as a last resort for refractory cases that cannot be controlled even after multiple surgeries 6).
When corneal endothelial damage progresses and corneal opacity becomes severe, corneal endothelial transplantation is indicated.
DSAEK/DSEK: Has a track record for corneal decompensation due to ICE syndrome 1). Graft survival is equivalent to penetrating keratoplasty (PKP), but visual recovery is faster and astigmatism is milder 1).
DMEK (Descemet membrane endothelial keratoplasty): There are reports of DMEK performed for ICE syndrome 7). Surgical technique requires placing the main incision to avoid PAS and using a smaller 7.5 mm donor tissue to bypass the PAS area. Fixation is achieved with 20% sulfur hexafluoride (SF6) gas, and postoperative intraocular pressure is managed with acetazolamide SR 250 mg for 2 days 7).
Saleki et al. (2025) published the first report of DMEK performed for ICE syndrome 7). DMEK was performed on the left eye of a 60-year-old man, and uncorrected visual acuity improved from preoperative 1.1 LogMAR to 0.54 LogMAR at 2 months and 0.4 LogMAR at 10 months postoperatively. Intraocular pressure stabilized at 16 mmHg, and corneal clarity was maintained up to 18 months.
PKP (penetrating keratoplasty): Indications are limited because maintaining corneal graft clarity is difficult in cases with poor intraocular pressure control.
A two-stage surgical approach has also been reported: phacoemulsification with artificial iris implantation followed by DSAEK 6 months later 1). In a 54-year-old woman, postoperative corrected visual acuity improved from 20/100 to 20/32, and corneal endothelial cell density was maintained at 1,640 cells/mm² 1).
Cogan-Reese syndrome is rarely associated with cystoid macular edema (CME) 3). Treatment with topical NSAIDs (flurbiprofen 3 times daily) led to resolution, but recurrence after discontinuation has been reported 3).
QCan corneal transplantation and tube shunt surgery be performed simultaneously?
A
Simultaneous surgery is possible, but a staged approach of intraocular pressure control followed by corneal transplantation has been reported in ICE syndrome. Stabilizing intraocular pressure before performing corneal endothelial transplantation is expected to improve graft survival. The optimal timing of glaucoma surgery and corneal transplantation should be determined on a case-by-case basis 1).
The fundamental abnormality in ICE syndrome is epithelialization of the corneal endothelium. Normal corneal endothelial cells are non-proliferative monolayer cells, but in ICE syndrome, they are replaced by epithelial-like cells with proliferative and migratory capacity. This process is also common to PPMD, but ICE is unilateral and sporadic, whereas PPMD is bilateral and autosomal dominant.
When an abnormality occurs in the corneal endothelium, a membranous tissue proliferates from the posterior corneal surface to the angle and anterior iris, extending beyond Schwalbe’s line onto the trabecular meshwork and further onto the anterior iris5). Contraction of this membranous tissue leads to the following pathologies.
The main mechanism is obstruction of aqueous humor outflow due to formation of tall peripheral anterior synechiae (PAS) 5). In addition, the membranous tissue itself may functionally close the trabecular meshwork, and glaucoma can occur even without obvious PAS. Resistance to medical treatment is due to the continuous progression of the membranous tissue.
Two mechanisms are involved: pump dysfunction of degenerated endothelial cells and elevated intraocular pressure due to glaucoma. In Chandler syndrome, endothelial cell damage is prominent, and corneal edema can occur even when intraocular pressure is within the normal range. When corneal endothelial cell density falls below 500 cells/mm², bullous keratopathy develops.
Contraction of the membranous tissue pulls the iris, causing pupillary deviation, iris hole formation, and ectropion uveae. This is most pronounced in progressive iris atrophy. Pupillary deviation occurs toward the direction with more PAS, and iris holes occur frequently in the opposite direction.
Histologically, iris nodules consist of melanin-containing spindle-shaped nevus cells, with a Ki-67 positivity rate of less than 1% and melan-A positivity, indicating a benign lesion 4). Rarely, they are associated with zonular fragility, and diffuse zonular dialysis may be observed during cataract surgery 4).
Immunohistochemistry of Descemet’s membrane excised from ICE syndrome eyes shows a mixed expression pattern of epithelial markers (AE1/AE3, CK8/18) and endothelial markers (CD56, vimentin), supporting that epithelial metaplasia is the essential pathology of ICE syndrome 7). Differentiation from epithelial downgrowth is important, but it is determined by a combination of clinical course and histological findings.
QHow is ICE syndrome differentiated from posterior polymorphous corneal dystrophy (PPMD)?
A
The simplest differentiating point is that ICE syndrome is sporadic and unilateral, whereas PPMD is autosomal dominant and bilateral. Specular microscopy also differs: ICE syndrome shows dark areas with central highlights (ICE cells), while PPMD presents typical vesicles or band-like structures. PPMD may also show iris synechiae and corneal edema, but it is congenital with no gender difference, so the clinical course differs.
Pinheiro-Costa J, Maia J, Branco A, et al. Two-step iridocorneal endothelial syndrome management: endocapsular intraocular lens implantation and Descemet’s stripping automated endothelial keratoplasty. Case Rep Ophthalmol. 2023;14(1):478-486.
Guler Canozer D, Unlu M, Gultekin Irez B, Ozkurt Y. In vivo confocal microscopy and anterior segment optical coherence tomography findings in iridocorneal endothelial syndrome. Turk J Ophthalmol. 2024;54(5):325-330.
Bouvarel H, Hamard P, Agard E, Billant J, El Chehab H, Dot C. Macular edema in Cogan-Reese syndrome. American journal of ophthalmology case reports. 2022;25:101318. doi:10.1016/j.ajoc.2022.101318. PMID:35128161; PMCID:PMC8810354.
Chhadva P, Del Valle Estopinal M, Farid M. Iris Nevus (Cogan-Reese) Syndrome Presenting with Zonular Dehiscence during Cataract Extraction. Case reports in ophthalmology. 2022;13(2):435-440. doi:10.1159/000524823. PMID:35950024; PMCID:PMC9247537.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Saleki M, Lee P, Thaung C, Ashena Z. Descemet’s membrane endothelial keratoplasty in an eye with iridocorneal endothelial syndrome and rare association of corneal ectasia. Ther Adv Ophthalmol. 2025;17:25158414251343968.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.