Le syndrome ICE (syndrome endothélial iridocornéen) est une maladie dans laquelle des cellules endothéliales cornéennes anormales s’étendent dans l’angle et l’iris avec un tissu membraneux, obstruant le trabéculum et provoquant une augmentation de la pression intraoculaire. L’anomalie de l’endothélium cornéen provoque un œdème cornéen, et la contraction du tissu membraneux entraîne des anomalies de l’iris et des synéchies antérieures périphériques (PAS).
Elle survient unilatéralement chez les femmes dans la trentaine et la quarantaine2). Elle n’est pas héréditaire et est sporadique. Les antécédents familiaux sont rares et il n’y a pas d’association cohérente avec d’autres maladies oculaires ou systémiques.
Le syndrome ICE est classé en trois types cliniques selon l’état des anomalies de l’iris :
Atrophie progressive de l’iris (progressive iris atrophy) : L’atrophie de l’iris est marquée, caractérisée par une déviation pupillaire, un ectropion uvéal et de multiples trous iriens. C’est le type le plus fréquemment associé au glaucome.
Syndrome de Chandler : le plus fréquent (environ 50 %). L’œdème cornéen dû à une insuffisance endothéliale est prédominant, tandis que la déviation pupillaire et l’atrophie de l’iris sont légères. L’œdème cornéen survient facilement même si l’élévation de la pression intraoculaire est légère.
Syndrome de Cogan-Reese (syndrome du naevus irien) : caractérisé par des nodules pigmentés sur la face antérieure de l’iris, avec deux types : nodules pédiculés et lésions pigmentées planes4). C’est le sous-type le plus rare.
Le syndrome ICE est unilatéral et sporadique, ce qui constitue le point de différenciation le plus simple avec la dystrophie cornéenne postérieure polymorphe (PPMD) et le syndrome d’Axenfeld-Rieger, qui sont bilatéraux et à transmission autosomique dominante.
QComment distinguer les trois types du syndrome ICE ?
A
La distinction entre les trois types repose principalement sur les signes cliniques de l’iris et de la cornée. L’atrophie progressive de l’iris se caractérise par une polycorie, une déviation pupillaire, des trous iriens et une ectropion uvéal, avec la plus forte incidence de glaucome. Le syndrome de Chandler présente un œdème cornéen prédominant avec des modifications iriennes minimes, pas de trous iriens et une hauteur de PAS faible. Le syndrome de Cogan-Reese se caractérise par des nodules pédiculés ou des lésions pigmentées planes sur la face antérieure de l’iris, et l’atrophie de l’iris est généralement absente. La physiopathologie de base est commune à tous les types, et la stratégie thérapeutique est similaire.
La baisse de l’acuité visuelle unilatérale (vision trouble due à l’œdème cornéen) est le symptôme initial le plus fréquent. Une photophobie et des douleurs peuvent être ressenties. Certains patients consultent en raison d’anomalies d’apparence telles qu’une déviation pupillaire ou une hétérochromie irienne. La douleur due à l’œdème cornéen et celle due à l’élévation de la pression intraoculaire par fermeture de l’angle coexistent.
Signes cliniques (constatations observées par le médecin lors de l’examen)
Signes cornéens : Dans le syndrome de Chandler, on observe un aspect métallique brillant de l’endothélium cornéen, appelé « hammered silver appearance » (aspect d’argent martelé). Cela ressemble aux gouttes cornéennes de la dystrophie endothéliale de Fuchs, mais peut être différencié par le caractère unilatéral. L’œdème cornéen peut survenir même si la pression intraoculaire est normale. Des plis de la membrane de Descemet peuvent également être observés.
Signes iriens : Selon le type, les signes suivants apparaissent2).
Atrophie irienne progressive
Déviation pupillaire : La pupille est tirée dans la direction où les PAS sont nombreux.
Formation de trous iriens : Multiples dans la direction opposée à la traction pupillaire.
Éversion uvéale : L’épithélium pigmentaire irien est exposé à la surface.
PAS : Des PAS hauts s’étendant au-delà de la ligne de Schwalbe sont caractéristiques.
Glaucome associé : le plus fréquent des trois types.
Syndrome de Chandler
Œdème cornéen : survient facilement même avec une légère élévation de la pression intraoculaire, le plus marqué des trois types.
Signes iriens : déviation pupillaire et atrophie irienne légères. Pas de trous iriens.
PAS : de faible hauteur.
Aspect endothélial : aspect argenté martelé. Au spéculaire, anisocytose et atypie étendues.
Syndrome de Cogan-Reese
Nodules iriens : deux types sont observés : de petits nodules pédiculés et des lésions pigmentaires plates.
Caractéristiques histologiques : lésion bénigne composée de cellules naviformes fusiformes contenant de la mélanine4).
Éversion uvéale : peut être associée.
Diagnostic différentiel : une différenciation avec un mélanome malin de l’iris peut être nécessaire.
Signes gonioscopiques : à l’examen gonioscopique, on observe des PAS s’élevant haut au-delà de la ligne de Schwalbe, ce qui est caractéristique du syndrome ICE5). Les PAS sont en plaques, très antérieures, et le trabéculum entre les PAS semble normal5). La forme des PAS varie : en tente, trapézoïdale, ou en adhérences étendues.
La véritable étiologie du syndrome ICE n’est pas élucidée, mais on suppose qu’une infection latente par le virus de l’herpès simplex (HSV) provoque une inflammation de bas grade au niveau de l’endothélium cornéen, entraînant une activation de type épithélial. Des tests PCR ont rapporté la détection d’ADN du HSV dans plus de 60 % des échantillons de cornée et d’humeur aqueuse de patients ICE 2). L’implication du virus d’Epstein-Barr (EBV) est également suggérée.
Sur le plan pathologique, les cellules endothéliales normales sont remplacées par des cellules de type épithélial possédant des propriétés de migration. La microscopie électronique révèle des caractéristiques épithéliales telles que des desmosomes, des tonofilaments et des microvillosités. Des lésions toxiques (modifications nécrotiques) des cellules endothéliales normales adjacentes ont également été rapportées.
L’examen histopathologique de la membrane de Descemet réséquée d’yeux atteints du syndrome ICE montre une monocouche de cellules cuboïdes basses avec une stratification partielle en bicouche ou multicouche, et l’immunohistochimie confirme un profil d’expression mixte des marqueurs épithéliaux (AE1/AE3, CK8/18) et endothéliaux (CD56, vimentine)7). Cette observation soutient pathologiquement la métaplasie épithélioïde, essence du syndrome ICE.
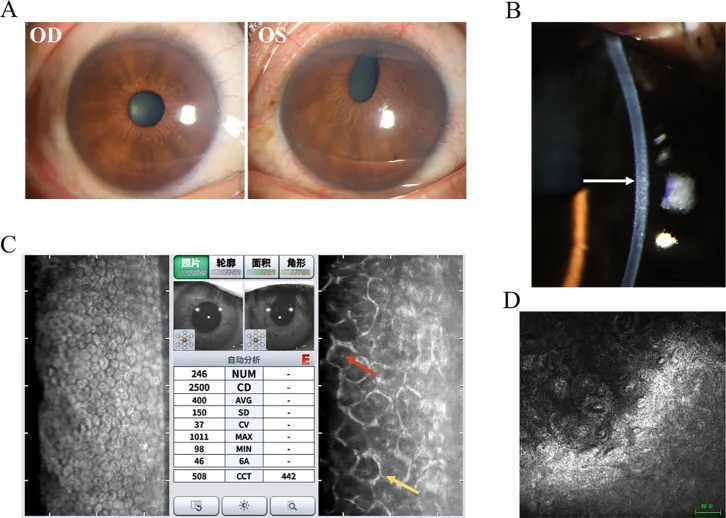
Hua Ma; Mingfang Xia; Qing Gu; et al. Iridocorneal endothelial syndrome. Front Ophthalmol (Lausanne). 2025;5:1655669. Figure 1. PMCID: PMC12537377. License: CC BY.
Figure résumant les signes représentatifs du syndrome de Chandler. Outre les photographies à la lampe à fente, les résultats d’examens complémentaires sont également présentés, permettant de visualiser facilement ce qu’il faut vérifier dans le syndrome ICE.
C’est l’examen le plus important pour le diagnostic définitif du syndrome ICE2). Le microscope spéculaire permet d’observer des cellules anormales (cellules ICE) présentant des zones sombres intracellulaires, ce qui permet le diagnostic. La mosaïque endothéliale hexagonale normale disparaît, montrant un pléomorphisme et un « renversement clair-obscur (light-dark reversal) ». Les cellules endothéliales sont caractéristiquement plus grandes et plus sombres que la normale, avec un point central brillant.
On observe une diminution marquée de la densité des cellules endothéliales cornéennes. La valeur normale est d’environ 3 500 cells/mm² chez les jeunes et de 2 500 à 3 000 cells/mm² chez les personnes âgées, mais lorsqu’elle descend en dessous de 500 cells/mm², la fonction de pompe est compromise, entraînant une kératopathie bulleuse. Dans un cas de syndrome de Cogan-Reese, une diminution à 763 cells/mm² a été rapportée 3).
Dans le syndrome de Chandler, l’examen spéculaire montre une anisocytose et une atypie étendues des cellules, et les limites des cellules endothéliales sont floues, ce qui aide au diagnostic. Un coefficient de variation (CV) supérieur à 0,35 est considéré comme anormal, et un pourcentage de cellules hexagonales inférieur à 50 % est également anormal.
Elle visualise des cellules endothéliales cornéennes gonflées en pavé, une disparition de la structure hexagonale, des cellules très brillantes polymorphes, et des cellules endothéliales géantes mononucléées ou binucléées 2). Elle est également utile lorsque l’évaluation par microscopie spéculaire est difficile en raison d’un œdème cornéen.
Il visualise les adhérences de l’angle iridocornéen, un épaississement hyperréfléchissant de la couche endothéliale cornéenne et un tissu membraneux sur l’iris2). Il est excellent pour l’évaluation quantitative de l’étendue et de la hauteur des PAS.
Elle est indispensable pour l’évaluation des PAS et est utilisée pour le diagnostic et le suivi du glaucome secondaire par fermeture de l’angle. Dans le syndrome ICE, les PAS dépassant la ligne de Schwalbe sont caractéristiques, et le trabéculum entre les PAS semble normal, ce qui est utile pour la différenciation du glaucome primitif à angle ouvert (POAG) 5).
La mesure de la pression intraoculaire, la photographie de la papille optique, le champ visuel (Humphrey ou Goldmann) et l’évaluation de l’épaisseur de la couche de fibres nerveuses rétiniennes (RNFL) par OCT sont introduits dans l’examen initial de précision et le suivi2).
Nécessite un diagnostic différentiel avec le syndrome de Cogan-Reese
QPourquoi le syndrome ICE est-il parfois diagnostiqué à tort comme un glaucome à angle ouvert ?
A
La membrane de cellules endothéliales cornéennes en progression peut fermer fonctionnellement le trabéculum sans contraction. Dans ce cas, l’absence de synéchies antérieures périphériques (PAS) évidentes à la gonioscopie conduit à un diagnostic erroné de glaucome à angle ouvert, malgré une fermeture angulaire « fonctionnelle ». En présence d’un glaucome unilatéral, le syndrome ICE doit être envisagé dans le diagnostic différentiel, et l’endothélium cornéen doit être soigneusement évalué par microscopie spéculaire.
Le traitement du syndrome ICE repose sur deux axes : le contrôle de la pression intraoculaire (PIO) du glaucome et la gestion de la décompensation cornéenne. Le contrôle à long terme de la PIO par traitement médicamenteux est souvent difficile, et une intervention chirurgicale est finalement nécessaire dans de nombreux cas.
Traitement médical
Inhibiteurs de la production d’humeur aqueuse : Les bêtabloquants topiques, les agonistes alpha-adrénergiques et les inhibiteurs de l’anhydrase carbonique (IAC) sont les traitements de première intention 6). La prescription suit celle du glaucome primitif à angle ouvert (GPAO). Les miotiques ciblant la voie d’écoulement de l’humeur aqueuse sont inefficaces et ne sont pas recommandés.
Analogues des prostaglandines : En raison de l’hypothèse d’une infection latente par le virus herpès simplex (HSV), il existe un risque de réactivation du HSV induite par le latanoprost, ce qui nécessite une évaluation prudente 2).
Prise en charge de l’œdème cornéen : Des collyres ou gels de solution saline hypertonique sont utilisés pour déshydrater la cornée.
Pronostic à long terme : En raison de la progression de la membrane hyaline qui élargit le PAS, le traitement médicamenteux devient souvent résistant 6).
Traitement chirurgical
Trabéculectomie : Réalisée avec un antifibrotique (mitomycine C ; MMC ou 5-FU) 6). Les taux de survie rapportés sont de 73 % à 1 an, 44 % à 3 ans et 29 % à 5 ans. Il existe un risque d’obstruction de la fistule par la membrane endothéliale anormale, et le taux de succès est faible.
Chirurgie de shunt tubulaire : La chirurgie de shunt tubulaire avec plaque est souvent choisie 5)6). Les taux de survie sont de 71 % à 1 an, 71 % à 3 ans et 53 % à 5 ans, ce qui est meilleur à long terme que la trabéculectomie.
Cyclodestruction : La photocoagulation cyclique au laser semi-conducteur (diode CPC) est envisagée comme dernier recours pour les cas réfractaires non contrôlés même après plusieurs chirurgies 6).
Lorsque la décompensation endothéliale cornéenne progresse et que l’opacité cornéenne devient sévère, une greffe endothéliale cornéenne est indiquée.
DSAEK/DSEK : A fait ses preuves dans la décompensation cornéenne due au syndrome ICE1). Le taux de survie du greffon est équivalent à celui de la kératoplastie transfixiante (PKP), mais la récupération visuelle est plus rapide et l’astigmatisme est moindre 1).
DMEK (greffe de membrane de Descemet) : Des rapports de DMEK pour le syndrome ICE existent 7). Des précautions techniques sont nécessaires, comme placer l’incision principale pour éviter les PAS et utiliser un greffon donneur plus petit de 7,5 mm pour contourner la zone PAS. Le greffon est fixé avec du gaz hexafluorure de soufre (SF6) à 20 %, et l’acétazolamide SR 250 mg est administré pendant 2 jours en postopératoire pour gérer la pression intraoculaire7).
Saleki et al. (2025) ont publié le premier rapport de DMEK pour le syndrome ICE7). Une DMEK a été réalisée sur l’œil gauche d’un homme de 60 ans. L’acuité visuelle non corrigée est passée de 1,1 LogMAR en préopératoire à 0,54 LogMAR à 2 mois postopératoires et à 0,4 LogMAR à 10 mois. La pression intraoculaire était stable à 16 mmHg et la transparence cornéenne a été maintenue jusqu’à 18 mois.
PKP (kératoplastie transfixiante) : Les indications sont limitées car le maintien de la transparence du greffon est difficile en cas de mauvais contrôle de la pression intraoculaire.
Une approche chirurgicale en deux étapes a également été rapportée : phacoémulsification + implantation d’iris artificiel en premier, suivie d’une DSAEK 6 mois plus tard 1). Chez une femme de 54 ans, l’acuité visuelle corrigée est passée de 20/100 à 20/32, et la densité des cellules endothéliales cornéennes de 1 640 cellules/mm² a été maintenue 1).
Le syndrome de Cogan-Reese peut rarement être associé à un œdème maculaire cystoïde (OMC) 3). Un traitement par AINS locaux (flurbiprofène 3 fois par jour) a permis la résolution, mais une récidive a été rapportée après l’arrêt 3).
QPeut-on réaliser simultanément une greffe de cornée et une chirurgie de tube de drainage ?
A
Une réalisation simultanée est possible, mais dans le syndrome ICE, une approche par étapes du contrôle de la pression intraoculaire et de la greffe de cornée a été rapportée. En stabilisant d’abord la pression intraoculaire puis en effectuant une greffe endothéliale, on espère améliorer la survie du greffon. Le moment optimal pour la chirurgie du glaucome et la greffe de cornée est déterminé en fonction de chaque cas 1).
L’anomalie fondamentale du syndrome ICE réside dans la métaplasie épithéliale (épithélialisation) de l’endothélium cornéen. Les cellules endothéliales cornéennes normales sont des cellules monocouches non prolifératives, mais dans le syndrome ICE, elles sont remplacées par des cellules de type épithélial ayant une capacité de prolifération et de migration. Ce processus est commun à la PPMD, mais l’ICE est unilatérale et sporadique, tandis que la PPMD est bilatérale et autosomique dominante.
Lorsque l’endothélium cornéen devient anormal, un tissu membraneux se développe de la face postérieure de la cornée vers l’angle et la face antérieure de l’iris, dépassant la ligne de Schwalbe pour s’étendre sur le trabéculum et la face antérieure de l’iris5). La contraction de ce tissu membraneux entraîne les pathologies suivantes.
Le mécanisme principal est l’obstruction de la voie d’écoulement de l’humeur aqueuse par la formation de synéchies antérieures périphériques (PAS) hautes 5). De plus, le tissu membraneux lui-même peut fermer fonctionnellement le trabéculum, et un glaucome peut survenir même en l’absence de PAS évidentes. La résistance au traitement médicamenteux est due à la progression continue du tissu membraneux.
Deux mécanismes sont impliqués : le dysfonctionnement de la pompe des cellules endothéliales dégénérées et l’augmentation de la pression intraoculaire due au glaucome. Dans le syndrome de Chandler, l’atteinte des cellules endothéliales est marquée et un œdème cornéen peut survenir même si la pression intraoculaire est normale. Lorsque la densité des cellules endothéliales cornéennes tombe en dessous de 500 cellules/mm², une kératopathie bulleuse se développe.
La contraction du tissu membraneux entraîne une traction de l’iris, provoquant une déviation pupillaire, une formation de trous iriens et une ectropion uvéal. Cela est le plus marqué dans l’atrophie irienne progressive. La déviation pupillaire se produit dans la direction où les PAS sont nombreuses, et les trous iriens apparaissent fréquemment dans la direction opposée.
Histologiquement, les nodules iriens sont constitués de cellules naviformes bénignes contenant de la mélanine, avec un taux de positivité Ki-67 inférieur à 1 % et une positivité melan-A, ce qui en fait des lésions bénignes 4). Rarement, ils peuvent être associés à une fragilité zonulaire, et une dialyse zonulaire diffuse peut être observée lors de la chirurgie de la cataracte4).
L’immunohistochimie de la membrane de Descemet retirée d’yeux atteints du syndrome ICE montre un motif d’expression mixte de marqueurs épithéliaux (AE1/AE3, CK8/18) et endothéliaux (CD56, vimentine), confirmant que la métaplasie épithéliale est la pathologie essentielle du syndrome ICE7). La distinction avec la prolifération épithéliale descendante (down-growth épithélial) est importante, mais elle se fait par la combinaison de l’évolution clinique et des résultats histologiques.
QComment distinguer le syndrome ICE de la dystrophie cornéenne postérieure polymorphe (PPMD) ?
A
Le point de distinction le plus simple est que le syndrome ICE est sporadique et unilatéral, tandis que la PPMD est autosomique dominante et bilatérale. La microscopie spéculaire diffère également : dans le syndrome ICE, on observe des zones sombres avec un reflet central (cellules ICE), alors que la PPMD présente des vésicules typiques ou des structures en bandes. La PPMD peut également montrer des synéchies antérieures de l’iris ou un œdème cornéen, mais comme elle est congénitale et sans différence de sexe, son évolution clinique est différente.
Pinheiro-Costa J, Maia J, Branco A, et al. Two-step iridocorneal endothelial syndrome management: endocapsular intraocular lens implantation and Descemet’s stripping automated endothelial keratoplasty. Case Rep Ophthalmol. 2023;14(1):478-486.
Guler Canozer D, Unlu M, Gultekin Irez B, Ozkurt Y. In vivo confocal microscopy and anterior segment optical coherence tomography findings in iridocorneal endothelial syndrome. Turk J Ophthalmol. 2024;54(5):325-330.
Bouvarel H, Hamard P, Agard E, Billant J, El Chehab H, Dot C. Macular edema in Cogan-Reese syndrome. American journal of ophthalmology case reports. 2022;25:101318. doi:10.1016/j.ajoc.2022.101318. PMID:35128161; PMCID:PMC8810354.
Chhadva P, Del Valle Estopinal M, Farid M. Iris Nevus (Cogan-Reese) Syndrome Presenting with Zonular Dehiscence during Cataract Extraction. Case reports in ophthalmology. 2022;13(2):435-440. doi:10.1159/000524823. PMID:35950024; PMCID:PMC9247537.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Saleki M, Lee P, Thaung C, Ashena Z. Descemet’s membrane endothelial keratoplasty in an eye with iridocorneal endothelial syndrome and rare association of corneal ectasia. Ther Adv Ophthalmol. 2025;17:25158414251343968.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.