Das ICE-Syndrom (Iridokorneales Endothelsyndrom) ist eine Erkrankung, bei der abnormale Hornhautendothelzellen mit einer membranartigen Struktur in den Kammerwinkel und die Iris einwachsen, das Trabekelwerk verstopfen und einen erhöhten Augeninnendruck verursachen. Die Anomalie des Hornhautendothels führt zu Hornhautödem, und die Kontraktion der membranartigen Struktur führt zu Irisanomalien und peripheren anterioren Synechien (PAS).
Sie tritt einseitig bevorzugt bei Frauen im Alter von 30–40 Jahren auf 2). Sie ist nicht erblich und tritt sporadisch auf. Eine Familienanamnese ist selten, und es gibt keinen konsistenten Zusammenhang mit anderen Augenerkrankungen oder systemischen Erkrankungen.
Das ICE-Syndrom wird je nach Zustand der Irisanomalie in die folgenden drei klinischen Typen eingeteilt:
Progressive Irisatrophie (progressive iris atrophy): Die Irisatrophie ist ausgeprägt, gekennzeichnet durch Pupillenverlagerung, Eversion des Uveapigmentepithels und multiple Iriskolobome. Dies ist der Typ mit der häufigsten Glaukomkomorbidität.
Chandler-Syndrom: Am häufigsten (ca. 50%). Hauptmerkmal ist das Hornhautödem aufgrund einer endothelialen Dysfunktion, während Pupillenverlagerung und Irisatrophie mild sind. Selbst bei leichtem Augeninnendruckanstieg tritt leicht ein Hornhautödem auf.
Cogan-Reese-Syndrom (Iris-Nävus-Syndrom): Gekennzeichnet durch pigmentierte Knoten auf der Irisvorderfläche, wobei zwei Arten unterschieden werden: gestielte Knoten und flache Pigmentherde 4). Es ist die seltenste Unterform.
Das ICE-Syndrom ist einseitig und sporadisch, was den einfachsten Unterscheidungspunkt zur beidseitigen, autosomal-dominant vererbten hinteren polymorphen Hornhautdystrophie (PPMD) und zum Axenfeld-Rieger-Syndrom darstellt.
QWie werden die drei Typen des ICE-Syndroms unterschieden?
A
Die Unterscheidung der drei Typen basiert hauptsächlich auf den klinischen Befunden von Iris und Hornhaut. Die progressive Irisatrophie ist durch Polykorie, Pupillenverlagerung, Iriskolobom und Eversion des Uveapigmentblattes gekennzeichnet und weist die häufigste Glaukomkomorbidität auf. Das Chandler-Syndrom zeigt vorwiegend ein Hornhautödem mit geringfügigen Irisveränderungen, keine Iriskolobome und niedrige PAS. Das Cogan-Reese-Syndrom ist durch gestielte Knoten oder flache Pigmentherde auf der Irisvorderfläche charakterisiert, eine Irisatrophie tritt in der Regel nicht auf. Allen Typen liegt die gleiche grundlegende Pathophysiologie zugrunde, und die Behandlungsstrategie ist ähnlich.
Das häufigste Erstsymptom ist eine einseitige Sehverschlechterung (Nebelsehen aufgrund von Hornhautödem). Es können Photophobie und Schmerzen auftreten. Manchmal wird der Arzt aufgrund äußerer Auffälligkeiten wie Pupillenverlagerung oder Iris-Heterochromie aufgesucht. Die Schmerzen können sowohl durch das Hornhautödem als auch durch den erhöhten Augeninnendruck infolge eines Kammerwinkelverschlusses verursacht werden.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Hornhautbefunde: Beim Chandler-Syndrom zeigt das Hornhautendothel ein metallisch glänzendes „hammered silver appearance“ (gehämmertes silberartiges Aussehen). Es ähnelt den Cornea guttata der Fuchs-Endotheldystrophie, ist aber durch die Einseitigkeit unterscheidbar. Ein Hornhautödem kann auch bei normalem Augeninnendruck auftreten. Es können auch Falten in der Descemet-Membran auftreten.
Irisbefunde: Je nach Typ treten folgende Befunde auf2):
Progressive Irisatrophie
Pupillenverziehung: Die Pupille wird in Richtung der vermehrten PAS gezogen.
Irislochbildung: Mehrere Löcher entstehen in der entgegengesetzten Richtung der Pupillenverziehung.
Uveale Eversion: Das Irispigmentepithel wird an der Oberfläche freigelegt.
PAS: Charakteristisch sind hohe PAS, die über die Schwalbe-Linie hinausragen.
Glaukomkomorbidität: Am häufigsten von den drei Typen.
Chandler-Syndrom
Hornhautödem: Tritt auch bei leicht erhöhtem Augeninnendruck leicht auf, am ausgeprägtesten von den drei Typen.
Irisbefunde: Pupillenverlagerung und Irisatrophie sind mild. Keine Iriskolobome.
PAS: Niedrige Höhe.
Endothelbefunde: Gehämmertes Silberaussehen. Im Spiegelmikroskop weit verbreitete Anisozytose und Pleomorphie.
Cogan-Reese-Syndrom
Irisknoten: Es werden zwei Arten beobachtet: gestielte kleine Knötchen und flache Pigmentherde.
Histologischer Befund: Gutartige Läsion, bestehend aus melanozytenhaltigen spindelförmigen Nävuszellen4).
Uveale Eversion: Kann als Begleitbefund auftreten.
Differenzialdiagnose: Eine Abgrenzung zum malignen Iris-Melanom kann erforderlich sein.
Kammerwinkelbefund: In der Gonioskopie zeigen sich PAS, die über die Schwalbe-Linie hinaus nach oben reichen, ein charakteristischer Befund des ICE-Syndroms5). Die PAS sind fleckförmig und liegen sehr weit vorne; das Trabekelwerk zwischen den PAS erscheint normal5). Die Form der PAS variiert von zeltartig über trapezförmig bis hin zu ausgedehnten Verwachsungen.
Die wahre Ätiologie des ICE-Syndroms ist ungeklärt, aber es gibt die Hypothese, dass eine latente Infektion mit dem Herpes-simplex-Virus (HSV) auf der Ebene des Hornhautendothels eine geringgradige Entzündung auslöst und eine epithelartige Aktivierung verursacht. PCR-Tests haben berichtet, dass HSV-DNA in über 60 % der Hornhaut- und Kammerwasserproben von ICE-Patienten nachgewiesen wurde 2). Auch eine Beteiligung des Epstein-Barr-Virus (EBV) wird vermutet.
Pathologisch werden normale Endothelzellen durch epithelartige Zellen mit Migrationseigenschaften ersetzt. Elektronenmikroskopisch werden epitheliale Merkmale wie Desmosomen, Tonofilamente und Mikrovilli bestätigt. Auch toxische Schäden (nekrotische Veränderungen) an benachbarten normalen Endothelzellen wurden berichtet.
Bei der histopathologischen Untersuchung der Descemet-Membran, die von Augen mit ICE-Syndrom entfernt wurde, zeigt sich eine einschichtige Struktur niedriger kubischer Zellen mit teilweiser zwei- oder mehrschichtiger Anordnung. Immunhistochemisch wurde ein gemischtes Expressionsmuster epithelialer Marker (AE1/AE3, CK8/18) und endothelialer Marker (CD56, Vimentin) nachgewiesen 7). Dieser Befund untermauert pathologisch die epitheliale Metaplasie, die das Wesen des ICE-Syndroms darstellt.
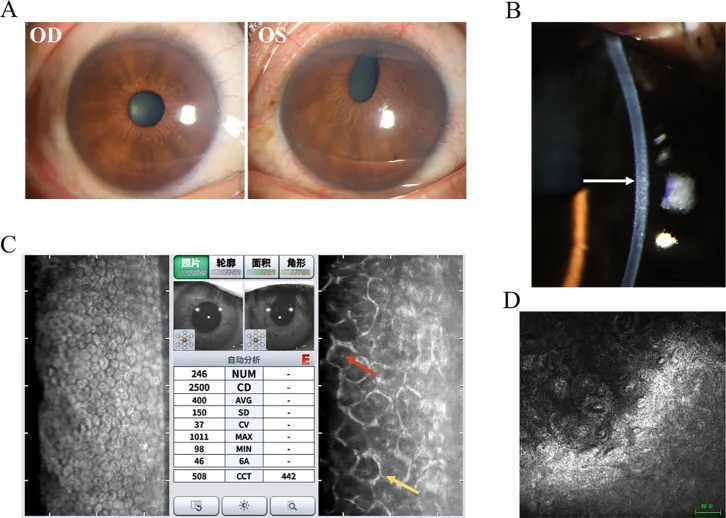
Hua Ma; Mingfang Xia; Qing Gu; et al. Iridocorneal endothelial syndrome. Front Ophthalmol (Lausanne). 2025;5:1655669. Figure 1. PMCID: PMC12537377. License: CC BY.
Diese Abbildung fasst die typischen Befunde des Chandler-Syndroms zusammen. Neben Spaltlampenfotos sind auch ergänzende Untersuchungsbefunde dargestellt, sodass leicht zu überblicken ist, was beim ICE-Syndrom überprüft wird.
Dies ist die wichtigste Untersuchung zur definitiven Diagnose des ICE-Syndroms 2). Mit dem Spiegelmikroskop werden abnormale Zellen (ICE-Zellen) mit dunklen Arealen im Zellinneren beobachtet, um die Diagnose zu stellen. Das normale sechseckige Endothelmosaik verschwindet, es zeigt sich Polymorphie und eine „Hell-Dunkel-Umkehr“ (light-dark reversal). Charakteristisch sind größere, dunklere Endothelzellen mit einem hellen Punkt in der Mitte.
Es zeigt sich eine deutliche Abnahme der Hornhautendothelzelldichte. Der Normalwert beträgt bei jungen Menschen etwa 3.500 Zellen/mm², bei älteren Menschen 2.500–3.000 Zellen/mm². Sinkt die Dichte unter 500 Zellen/mm², versagt die Pumpfunktion und es kommt zur bullösen Keratopathie. In einem Fall des Cogan-Reese-Syndroms wurde ein Rückgang auf 763 Zellen/mm² berichtet 3).
Beim Chandler-Syndrom zeigt die Spiegelmikroskopie ausgedehnte Anisozytose und Pleomorphismus der Zellen, und die Grenzen der Endothelzellen sind undeutlich, was zur Diagnose beiträgt. Ein Variationskoeffizient (CV) von ≥ 0,35 gilt als abnormal, und eine Hexagonalzellrate von ≤ 50 % wird als abnormal angesehen.
Sie stellt kopfsteinpflasterartig geschwollene Endothelzellen, Verlust der hexagonalen Struktur, polymorph hochreflektive Zellen sowie einkernige und zweikernige Riesenzellen des Endothels dar 2). Sie ist auch nützlich, wenn die Spiegelmikroskopie aufgrund eines Hornhautödems schwierig zu beurteilen ist.
Es visualisiert Verwachsungen des Kammerwinkels, eine hyperreflektive Verdickung der Hornhautendothelschicht und membranartiges Gewebe auf der Iris2). Es eignet sich hervorragend zur quantitativen Beurteilung des Ausmaßes und der Höhe von PAS.
Sie ist für die Beurteilung von PAS unerlässlich und wird zur Diagnose und Verlaufskontrolle des sekundären Winkelblockglaukoms eingesetzt. Beim ICE-Syndrom sind PAS, die über die Schwalbe-Linie hinausgehen, charakteristisch, und das Trabekelwerk zwischen den PAS erscheint normal, was zur Unterscheidung vom POAG nützlich ist 5).
Augeninnendruckmessung, Papillenfotografie, Gesichtsfelduntersuchung (Humphrey oder Goldmann) und OCT zur Beurteilung der retinalen Nervenfaserschichtdicke (RNFL) werden in die initiale Feindiagnostik und Verlaufskontrolle einbezogen 2).
QWarum wird das ICE-Syndrom manchmal fälschlicherweise als Offenwinkelglaukom diagnostiziert?
A
Eine vorrückende Hornhautendothelzellmembran kann das Trabekelwerk funktionell verschließen, ohne dass eine Kontraktion auftritt. In diesem Fall wird bei der Gonioskopie keine offensichtliche periphere anteriore Synechie (PAS) sichtbar, sodass trotz eines „funktionellen“ Kammerwinkelverschlusses fälschlicherweise ein Offenwinkelglaukom angenommen wird. Bei einem einseitigen Glaukom sollte das ICE-Syndrom in die Differenzialdiagnose einbezogen und das Hornhautendothel sorgfältig mittels Spiegelmikroskopie beurteilt werden.
Die Behandlung des ICE-Syndroms umfasst zwei Hauptaspekte: die Kontrolle des Augeninnendrucks bei Glaukom und die Behandlung der Hornhautdekompensation. Eine langfristige medikamentöse Drucksenkung ist oft schwierig, sodass viele Fälle letztendlich einen chirurgischen Eingriff erfordern.
Medikamentöse Therapie
Kammerwasserproduktionshemmer: Topische Betablocker, Alpha-Agonisten und Carboanhydrasehemmer (CAH) sind die erste Wahl 6). Die Verordnung erfolgt analog zum primären Offenwinkelglaukom (POAG). Miotika, die auf den Kammerwasserabfluss abzielen, sind wenig wirksam und werden nicht empfohlen.
Prostaglandinanaloga: Aufgrund der Hypothese einer latenten HSV-Infektion wird eine durch Latanoprost ausgelöste HSV-Reaktivierung befürchtet, daher ist eine sorgfältige Abwägung erforderlich 2).
Management von Hornhautödem: Hypertonische Kochsalzlösung als Augentropfen oder Gel wird zur Dehydrierung der Hornhaut eingesetzt.
Langzeitprognose: Da sich die PAS durch das Fortschreiten des membranösen Gewebes ausdehnt, spricht die medikamentöse Therapie oft nicht an 6).
Chirurgische Behandlung
Trabekulektomie: Wird in Kombination mit einem Antifibrotikum (Mitomycin C; MMC oder 5-FU) durchgeführt 6). Die Überlebensraten werden mit 73 % nach einem Jahr, 44 % nach drei Jahren und 29 % nach fünf Jahren angegeben. Es besteht das Risiko eines Fistelverschlusses durch die abnorme Endothelmembran, und die Erfolgsrate ist niedrig.
Tube-Shunt-Operation: Häufig wird eine Tube-Shunt-Operation mit Platte gewählt 5)6). Die Überlebensraten liegen bei 71 % nach einem Jahr, 71 % nach drei Jahren und 53 % nach fünf Jahren, was bessere Langzeitergebnisse als die Trabekulektomie zeigt.
Zyklodestruktive Verfahren: Die Diodenlaser-Zyklophotokoagulation (Dioden-CPC) wird als letzte Option für therapierefraktäre Fälle in Betracht gezogen, die auch nach mehreren Operationen nicht kontrollierbar sind 6).
Wenn die Hornhautendothelschädigung fortschreitet und die Hornhauttrübung schwerwiegend wird, ist eine Endothelkeratoplastik indiziert.
DSAEK/DSEK: Es gibt Erfahrungen mit der Behandlung der Hornhautdekompensation beim ICE-Syndrom1). Die Transplantatüberlebensrate ist vergleichbar mit der perforierenden Keratoplastik (PKP), jedoch ist die visuelle Erholung schneller und der Astigmatismus geringer 1).
DMEK (Descemet-Membran-Endothel-Keratoplastik): Es gibt Berichte über die Durchführung einer DMEK bei ICE-Syndrom7). Der Hauptschnitt sollte so platziert werden, dass PAS vermieden werden, und es sind technische Anpassungen erforderlich, um den PAS-Bereich mit einem kleineren 7,5-mm-Spendergewebe zu umgehen. Die Fixierung erfolgt mit 20% Schwefelhexafluorid (SF6)-Gas, und postoperativ wird Acetazolamid SR 250 mg für 2 Tage zur Augeninnendruckkontrolle verabreicht 7).
Saleki et al. (2025) veröffentlichten den ersten Bericht über eine DMEK bei ICE-Syndrom7). Bei einem 60-jährigen Mann wurde am linken Auge eine DMEK durchgeführt; der unkorrigierte Visus verbesserte sich von präoperativ 1,1 LogMAR auf 0,54 LogMAR nach 2 Monaten und 0,4 LogMAR nach 10 Monaten. Der Augeninnendruck war mit 16 mmHg stabil, und die Hornhauttransparenz blieb bis zu 18 Monate erhalten.
PKP (perforierende Keratoplastik): Bei schlecht kontrolliertem Augeninnendruck ist die Aufrechterhaltung der Hornhauttransparenz schwierig, daher sind die Indikationen eingeschränkt.
Als zweistufiger chirurgischer Ansatz wurde auch die Durchführung einer Phakoemulsifikation mit Kunstiris-Implantation, gefolgt von einer DSAEK nach 6 Monaten, berichtet 1). Bei einer 54-jährigen Frau verbesserte sich der korrigierte Visus von 20/100 auf 20/32, und die Endothelzeldichte blieb bei 1.640 Zellen/mm² erhalten 1).
Das Cogan-Reese-Syndrom kann selten mit einem zystoiden Makulaödem (CME) einhergehen 3). Eine Behandlung mit topischen NSAR (Flurbiprofen 3-mal täglich) führte zur Rückbildung, jedoch wurde ein Rezidiv nach Absetzen berichtet 3).
QKönnen Hornhauttransplantation und Tubenshunt-Operation gleichzeitig durchgeführt werden?
A
Eine gleichzeitige Durchführung ist möglich, aber beim ICE-Syndrom wird ein schrittweiser Ansatz mit Augeninnendruckkontrolle und Hornhauttransplantation berichtet. Zuerst wird der Augeninnendruck stabilisiert, dann erfolgt die Hornhautendotheltransplantation, was die Überlebensrate des Transplantats verbessern kann. Der optimale Zeitpunkt für Glaukomoperation und Hornhauttransplantation wird je nach Einzelfall bestimmt 1).
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Die grundlegende Anomalie des ICE-Syndroms ist die epitheliale Metaplasie (Epithelialisierung) des Hornhautendothels. Normale Hornhautendothelzellen sind nicht-proliferative einschichtige Zellen, aber beim ICE-Syndrom werden sie durch epitheliale Zellen mit Proliferations- und Migrationsfähigkeit ersetzt. Dieser Prozess ist auch bei der PPMD gemeinsam, aber das ICE ist einseitig und sporadisch, während die PPMD beidseitig und autosomal-dominant vererbt wird.
Wenn eine Anomalie des Hornhautendothels auftritt, wächst membranöses Gewebe von der Rückseite der Hornhaut über den Kammerwinkel und die Irisvorderfläche, überschreitet die Schwalbe-Linie und erstreckt sich auf das Trabekelwerk und weiter auf die Irisvorderfläche 5). Die Kontraktion dieses membranösen Gewebes führt zu den folgenden pathologischen Zuständen.
Der Hauptmechanismus ist die Verlegung des Kammerwasserabflusses durch hohe PAS-Bildung 5). Zusätzlich kann das membranöse Gewebe selbst das Trabekelwerk funktionell verschließen, sodass ein Glaukom auch ohne offensichtliche PAS auftreten kann. Die Resistenz gegen medikamentöse Behandlung ist auf das Fortschreiten des membranösen Gewebes zurückzuführen.
Es sind zwei Mechanismen beteiligt: die Pumpfunktionsstörung der degenerierten Endothelzellen und der durch das Glaukom verursachte erhöhte Augeninnendruck. Beim Chandler-Syndrom ist die Endothelzellschädigung ausgeprägt, und ein Hornhautödem kann auch bei normalem Augeninnendruck auftreten. Wenn die Dichte der Hornhautendothelzellen auf unter 500 Zellen/mm² fällt, kommt es zur bullösen Keratopathie.
Durch die Kontraktion des membranösen Gewebes wird die Iris verzogen, was zu Pupillenverlagerung, Iriskolobom und Uveaeversion führt. Dies ist bei der progressiven Irisatrophie am ausgeprägtesten. Die Pupillenverlagerung erfolgt in Richtung der vermehrten PAS, und Iriskolobome treten gehäuft in der entgegengesetzten Richtung auf.
Histologisch bestehen die Irisknoten aus melaninhaltigen spindelförmigen Nävuszellen, mit einer Ki-67-Positivitätsrate von unter 1 % und Melan-A-Positivität, es handelt sich um gutartige Läsionen 4). Selten geht dies mit einer Zonulafragilität einher, sodass bei Kataraktoperationen eine diffuse Zonuladialyse auftreten kann 4).
Die Immunhistochemie der Descemet-Membran, die aus ICE-Syndrom-Augen entfernt wurde, zeigt ein gemischtes Expressionsmuster von epithelialen Markern (AE1/AE3, CK8/18) und endothelialen Markern (CD56, Vimentin), was die epitheliale Metaplasie als wesentliche Pathologie des ICE-Syndroms bestätigt 7). Die Abgrenzung zum epithelialen Down-Growth (epitheliale Invasion) ist wichtig, erfolgt jedoch durch die Kombination von klinischem Verlauf und histologischen Befunden.
QWie unterscheidet man das ICE-Syndrom von der hinteren polymorphen Hornhautdystrophie (PPMD)?
A
Der einfachste Unterscheidungspunkt ist, dass das ICE-Syndrom sporadisch und einseitig auftritt, während die PPMD autosomal-dominant vererbt wird und beidseitig ist. Auch die Spiegelmikroskopie unterscheidet sich: Beim ICE-Syndrom zeigen sich dunkle Bereiche mit zentralen Highlights (ICE-Zellen), während die PPMD typische Bläschen oder bandartige Strukturen aufweist. Die PPMD kann ebenfalls Irisvorderwandsynechien und Hornhautödem aufweisen, ist jedoch angeboren und zeigt keinen Geschlechtsunterschied, sodass der klinische Verlauf unterschiedlich ist.
Pinheiro-Costa J, Maia J, Branco A, et al. Two-step iridocorneal endothelial syndrome management: endocapsular intraocular lens implantation and Descemet’s stripping automated endothelial keratoplasty. Case Rep Ophthalmol. 2023;14(1):478-486.
Guler Canozer D, Unlu M, Gultekin Irez B, Ozkurt Y. In vivo confocal microscopy and anterior segment optical coherence tomography findings in iridocorneal endothelial syndrome. Turk J Ophthalmol. 2024;54(5):325-330.
Bouvarel H, Hamard P, Agard E, Billant J, El Chehab H, Dot C. Macular edema in Cogan-Reese syndrome. American journal of ophthalmology case reports. 2022;25:101318. doi:10.1016/j.ajoc.2022.101318. PMID:35128161; PMCID:PMC8810354.
Chhadva P, Del Valle Estopinal M, Farid M. Iris Nevus (Cogan-Reese) Syndrome Presenting with Zonular Dehiscence during Cataract Extraction. Case reports in ophthalmology. 2022;13(2):435-440. doi:10.1159/000524823. PMID:35950024; PMCID:PMC9247537.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Saleki M, Lee P, Thaung C, Ashena Z. Descemet’s membrane endothelial keratoplasty in an eye with iridocorneal endothelial syndrome and rare association of corneal ectasia. Ther Adv Ophthalmol. 2025;17:25158414251343968.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.