La sindrome ICE (sindrome endoteliale iridocorneale; iridocorneal endothelial syndrome) è una malattia in cui cellule anomale dell’endotelio corneale si estendono all’angolo e all’iride con tessuto membranoso, causando ostruzione del trabecolato e aumento della pressione intraoculare. L’anomalia dell’endotelio corneale provoca edema corneale, e la contrazione del tessuto membranoso causa anomalie dell’iride e aderenze periferiche anteriori dell’iride (PAS).
Si manifesta unilateralmente in donne tra i 30 e i 40 anni2). Non è ereditaria, ma sporadica. Una storia familiare è rara e non vi è una correlazione coerente con altre malattie oculari o sistemiche.
La sindrome ICE è classificata in tre tipi clinici in base allo stato dell’anomalia dell’iride.
Atrofia progressiva dell’iride (progressive iris atrophy): caratterizzata da atrofia marcata dell’iride, deviazione pupillare, ectropion uveale e formazione di fori multipli nell’iride. È il tipo con la più alta associazione di glaucoma.
Sindrome di Chandler: la più frequente (circa 50%). È caratterizzata principalmente da edema corneale dovuto a disfunzione endoteliale, mentre la deviazione pupillare e l’atrofia dell’iride sono lievi. L’edema corneale si verifica facilmente anche con un lieve aumento della pressione intraoculare.
Sindrome di Cogan-Reese (sindrome del nevo dell’iride): caratterizzata da noduli pigmentati sulla superficie anteriore dell’iride, di due tipi: noduli peduncolati e lesioni pigmentate piatte4). È il sottotipo più raro.
La distinzione dei tre tipi si basa principalmente sui reperti clinici dell’iride e della cornea. L’atrofia progressiva dell’iride presenta policoria, deviazione pupillare, fori dell’iride ed ectropion uveale marcati, ed è la più frequentemente associata a glaucoma. La sindrome di Chandler è caratterizzata principalmente da edema corneale con alterazioni iridee minime, nessun foro dell’iride e bassa altezza della membrana di PAS. La sindrome di Cogan-Reese è caratterizzata da noduli peduncolati o lesioni pigmentate piatte sulla superficie anteriore dell’iride, e di solito non si osserva atrofia dell’iride. Tutti i tipi condividono la stessa fisiopatologia di base e la strategia terapeutica è simile.
Il sintomo d’esordio più comune è la riduzione unilaterale dell’acuità visiva (visione offuscata dovuta a edema corneale). Possono verificarsi fotofobia e dolore. A volte i pazienti consultano un medico a causa di anomalie estetiche come deviazione pupillare o eterocromia dell’iride. Il dolore può essere dovuto sia all’edema corneale che all’aumento della pressione intraoculare da chiusura dell’angolo.
Reperti clinici (reperti osservati dal medico durante la visita)
Reperti corneali: Nella sindrome di Chandler, l’endotelio corneale presenta un aspetto metallico lucido, descritto come “hammered silver appearance” (aspetto d’argento martellato). È simile alle guttate corneali della distrofia endoteliale di Fuchs, ma si differenzia per la monolateralità. L’edema corneale può manifestarsi anche con pressione intraoculare normale. Possono essere presenti pieghe della membrana di Descemet.
Reperti iridei: A seconda del tipo, compaiono i seguenti reperti2).
Atrofia progressiva dell'iride
Deviazione pupillare: La pupilla è tirata nella direzione in cui sono più numerose le aderenze periferiche anteriori (PAS).
Formazione di fori iridei: Si verificano multipli fori nella direzione opposta alla trazione pupillare.
Eversione uveale: L’epitelio pigmentato dell’iride è esposto sulla superficie.
PAS: Sono caratteristiche le PAS alte che si estendono oltre la linea di Schwalbe.
Glaucoma associato: il più frequente tra i tre tipi.
Sindrome di Chandler
Edema corneale: si verifica facilmente anche con un lieve aumento della pressione intraoculare, è il più marcato tra i tre tipi.
Reperti iridei: la deviazione pupillare e l’atrofia dell’iride sono lievi. Non si osservano fori iridei.
PAS: di bassa altezza.
Reperti endoteliali: aspetto a martello d’argento. Alla speculare, anisocitosi e atipia estese.
Sindrome di Cogan-Reese
Noduli iridei: si osservano due tipi: piccoli noduli peduncolati e lesioni pigmentate piatte.
Diagnosi differenziale: può essere necessaria la differenziazione dal melanoma maligno dell’iride.
Reperti angolari: all’esame gonioscopico si osservano PAS che si estendono oltre la linea di Schwalbe, caratteristici della sindrome ICE5). Le PAS sono a chiazze e situate molto anteriormente; il trabecolato tra le PAS appare normale5). La forma delle PAS può variare: a tenda, trapezoidale o aderenze estese.
La vera eziologia della sindrome ICE non è ancora chiara, ma esiste l’ipotesi che un’infezione latente da virus herpes simplex (HSV) provochi un’infiammazione di basso grado a livello dell’endotelio corneale, inducendo un’attivazione simile a quella epiteliale. Studi di PCR hanno riportato il rilevamento di DNA di HSV in oltre il 60% dei campioni di cornea e umor acqueo di pazienti con ICE 2). È stato anche suggerito un coinvolgimento del virus di Epstein-Barr (EBV).
Patologicamente, le cellule endoteliali normali vengono sostituite da cellule simil-epiteliali con proprietà migratorie. Al microscopio elettronico si osservano caratteristiche epiteliali come desmosomi, tonofilamenti e microvilli. Sono stati riportati anche danni tossici (alterazioni necrotiche) alle cellule endoteliali normali adiacenti.
L’esame istopatologico della membrana di Descemet rimossa da occhi con sindrome ICE mostra uno strato singolo di cellule cuboidali basse con parziale stratificazione doppia o multipla, e l’immunoistochimica conferma un pattern di espressione mista di marcatori epiteliali (AE1/AE3, CK8/18) ed endoteliali (CD56, vimentina)7). Questo reperto supporta patologicamente la metaplasia epiteliale, che è l’essenza della sindrome ICE.
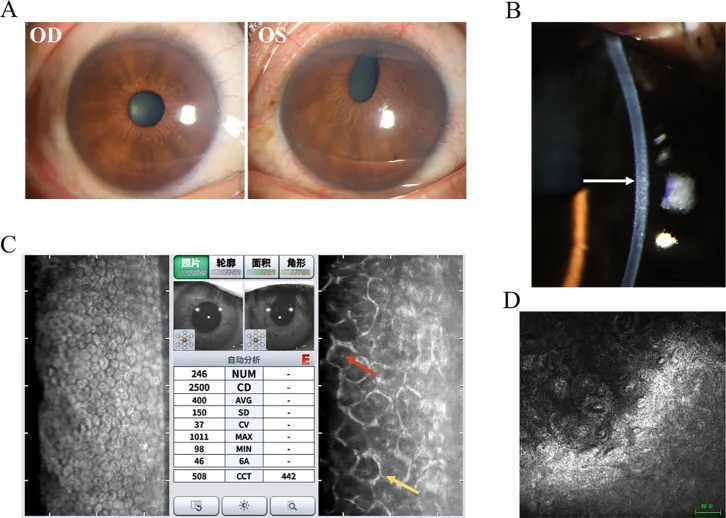
Hua Ma; Mingfang Xia; Qing Gu; et al. Iridocorneal endothelial syndrome. Front Ophthalmol (Lausanne). 2025;5:1655669. Figure 1. PMCID: PMC12537377. License: CC BY.
Figura che riassume i reperti tipici della sindrome di Chandler. Oltre alle fotografie con lampada a fessura, sono affiancati anche i reperti degli esami complementari, facilitando la comprensione complessiva di cosa verificare nella sindrome ICE.
È l’esame più importante per la diagnosi definitiva della sindrome ICE2). Con il microscopio speculare si osservano cellule anomale (cellule ICE) con aree scure all’interno della cellula, permettendo la diagnosi. Il normale mosaico endoteliale esagonale scompare, mostrando pleomorfismo e un ‘inversione chiaro-scuro’ (light-dark reversal). Sono caratteristiche cellule endoteliali più grandi e scure del normale con un punto centrale luminoso.
Si osserva una marcata riduzione della densità delle cellule endoteliali corneali. Il valore normale è di circa 3.500 cells/mm² nei giovani e di 2.500-3.000 cells/mm² negli anziani, ma quando scende al di sotto di 500 cells/mm², la funzione di pompa viene compromessa e si sviluppa la cheratopatia bollosa. In un caso di sindrome di Cogan-Reese è stato riportato un calo a 763 cells/mm² 3).
Nella sindrome di Chandler, l’esame speculare mostra un’ampia anisocitosi e atipia cellulare, con confini delle cellule endoteliali poco chiari, utile per la diagnosi. Il coefficiente di variazione (CV) è considerato anomalo se ≥ 0,35, e la percentuale di cellule esagonali è anomala se ≤ 50%.
Rivela cellule endoteliali corneali rigonfie a ciottolo, perdita della struttura esagonale, cellule iperriflettenti pleomorfe e cellule endoteliali giganti mononucleate o binucleate 2). È utile anche quando la microscopia speculare è difficile a causa dell’edema corneale.
Visualizza sinechie dell’angolo iridocorneale, ispessimento iperriflettente dello strato endoteliale corneale e tessuto membranoso sull’iride2). Eccellente per la valutazione quantitativa dell’estensione e dell’altezza delle PAS.
Essenziale per la valutazione delle PAS, utilizzata per la diagnosi e il follow-up del glaucoma secondario ad angolo chiuso. Nella sindrome ICE, le PAS che superano la linea di Schwalbe sono caratteristiche, e il fatto che il trabecolato tra le PAS appaia normale è utile per la diagnosi differenziale con il glaucoma primario ad angolo aperto5).
La misurazione della pressione intraoculare, la fotografia del disco ottico, il campo visivo (Humphrey o Goldmann) e la valutazione dello spessore dello strato di fibre nervose retiniche (RNFL) mediante OCT vengono introdotti nell’esame iniziale di precisione e nel follow-up2).
QPerché la sindrome ICE viene talvolta diagnosticata erroneamente come glaucoma ad angolo aperto?
A
La membrana delle cellule endoteliali corneali in avanzamento può chiudere funzionalmente il trabecolato senza contrazione. In questo caso, all’esame gonioscopico non si osservano evidenti PAS, quindi si può erroneamente diagnosticare un glaucoma ad angolo aperto nonostante sia presente una chiusura angolare “funzionale”. Quando si osserva un glaucoma unilaterale, si deve includere la sindrome ICE nella diagnosi differenziale e valutare attentamente l’endotelio corneale con la microscopia speculare.
Il trattamento della sindrome ICE si articola su due fronti: il controllo della pressione intraoculare nel glaucoma e la gestione del decompenso corneale. Spesso il controllo pressorio a lungo termine con terapia farmacologica risulta difficile e molti casi richiedono un intervento chirurgico.
Trattamento medico
Farmaci che sopprimono la produzione di umore acqueo: beta-bloccanti topici, agonisti alfa-adrenergici e inibitori dell’anidrasi carbonica (CAI) sono la prima scelta 6). La prescrizione segue quella per il glaucoma primario ad angolo aperto (POAG). I miotici che agiscono sulla via di deflusso dell’umore acqueo sono poco efficaci e non raccomandati.
Farmaci correlati alle prostaglandine: sulla base dell’ipotesi di infezione latente da HSV, esiste il timore di riattivazione dell’HSV indotta dal latanoprost, pertanto vanno usati con cautela 2).
Gestione dell’edema corneale: si utilizza una soluzione salina ipertonica in collirio o gel per disidratare la cornea.
Prognosi a lungo termine: poiché la progressione della membrana ialina porta all’espansione del PAS, il trattamento farmacologico diventa spesso resistente 6).
Trattamento chirurgico
Trabeculectomia: eseguita con l’uso di farmaci antifibrotici (mitomicina C; MMC o 5-FU) 6). I tassi di sopravvivenza riportati sono del 73% a 1 anno, 44% a 3 anni e 29% a 5 anni. Esiste il rischio di occlusione della fistola da parte della membrana endoteliale anomala, con conseguente basso tasso di successo.
Intervento di shunt tubulare: spesso si sceglie un intervento di shunt tubulare con placca 5)6). I tassi di sopravvivenza sono del 71% a 1 anno, 71% a 3 anni e 53% a 5 anni, risultati migliori a lungo termine rispetto alla trabeculectomia.
Ciclodistruzione: la fotocoagulazione cicliare con laser a semiconduttore (CPC a diodi) è considerata come ultima risorsa per i casi refrattari non controllabili nemmeno dopo multipli interventi 6).
Quando il danno endoteliale corneale progredisce e l’opacità corneale diventa grave, è indicato il trapianto di endotelio corneale.
DSAEK/DSEK: ha una comprovata efficacia per il compenso corneale nella sindrome ICE1). Ha un tasso di sopravvivenza dell’innesto simile al trapianto di cornea a tutto spessore (PKP), ma con un recupero visivo più rapido e un astigmatismo minore 1).
DMEK (trapianto di membrana di Descemet): Sono stati riportati casi di DMEK per la sindrome ICE7). È necessario posizionare l’incisione principale in modo da evitare le PAS e utilizzare un tessuto donatore più piccolo di 7,5 mm per evitare l’area PAS. Si fissa con gas esafluoruro di zolfo (SF6) al 20% e si somministra acetazolamide SR 250 mg per 2 giorni dopo l’intervento per gestire la pressione intraoculare7).
Saleki et al. (2025) hanno pubblicato il primo rapporto di DMEK eseguito per la sindrome ICE7). Un uomo di 60 anni è stato sottoposto a DMEK nell’occhio sinistro; l’acuità visiva non corretta è migliorata da 1,1 LogMAR preoperatorio a 0,54 LogMAR a 2 mesi e 0,4 LogMAR a 10 mesi dall’intervento. La pressione intraoculare si è stabilizzata a 16 mmHg e la trasparenza corneale è stata mantenuta fino a 18 mesi.
PKP (cheratoplastica perforante): Nei casi con scarso controllo della pressione intraoculare, è difficile mantenere la trasparenza dell’innesto corneale, quindi le indicazioni sono limitate.
Come approccio chirurgico in due fasi, è stato riportato un metodo che prevede prima la facoemulsificazione con impianto di iride artificiale, seguito da DSAEK dopo 6 mesi 1). In un caso di una donna di 54 anni, l’acuità visiva corretta è migliorata da 20/100 a 20/32 e la densità delle cellule endoteliali corneali è stata mantenuta a 1.640 cellule/mm² 1).
La sindrome di Cogan-Reese può raramente essere complicata da edema maculare cistoide (CME) 3). È stato riportato che il trattamento con FANS topici (flurbiprofene 3 volte al giorno) ha portato alla risoluzione, ma si è verificata una recidiva dopo la sospensione 3).
QÈ possibile eseguire contemporaneamente il trapianto di cornea e la chirurgia con tubo di drenaggio?
A
L’esecuzione simultanea è possibile, ma nella sindrome ICE è stato riportato un approccio graduale al controllo della pressione intraoculare e al trapianto di cornea. Stabilizzando prima la pressione intraoculare e poi eseguendo il trapianto di endotelio corneale, si prevede un miglioramento della sopravvivenza dell’innesto. Il momento ottimale per la chirurgia del glaucoma e il trapianto di cornea viene determinato caso per caso 1).
L’anomalia fondamentale nella sindrome ICE è la metaplasia epiteliale dell’endotelio corneale. Le normali cellule endoteliali corneali sono un monostrato non proliferativo, ma nella sindrome ICE vengono sostituite da cellule simili all’epitelio con capacità proliferativa e migratoria. Questo processo è comune anche alla PPMD, ma l’ICE è unilaterale e sporadico, mentre la PPMD è bilaterale e autosomica dominante.
Quando si verifica un’anomalia nell’endotelio corneale, un tessuto membranoso si propaga dalla superficie posteriore della cornea all’angolo e alla superficie anteriore dell’iride, superando la linea di Schwalbe, estendendosi sul trabecolato e quindi sulla superficie anteriore dell’iride5). La contrazione di questo tessuto membranoso provoca le seguenti condizioni patologiche.
Il meccanismo principale è l’ostruzione del deflusso dell’umore acqueo dovuta alla formazione di PAS alte 5). Inoltre, il tessuto membranoso stesso può chiudere funzionalmente il trabecolato, e il glaucoma può svilupparsi anche in assenza di PAS evidenti. La resistenza al trattamento farmacologico è dovuta alla progressione continua del tessuto membranoso.
Sono coinvolti due meccanismi: la disfunzione della pompa delle cellule endoteliali degenerate e l’aumento della pressione intraoculare dovuto al glaucoma. Nella sindrome di Chandler, il danno endoteliale è marcato e l’edema corneale può verificarsi anche con pressione intraoculare normale. Quando la densità delle cellule endoteliali corneali scende al di sotto di 500 cellule/mm², si arriva alla cheratopatia bollosa.
La contrazione del tessuto membranoso tira l’iride, causando deviazione pupillare, formazione di fori iridei ed ectropion uveale. È più marcata nell’atrofia progressiva dell’iride. La deviazione pupillare è diretta verso le aree con più PAS, mentre i fori iridei si verificano frequentemente nella direzione opposta.
I noduli iridei sono istologicamente composti da cellule neviche fusiformi contenenti melanina, con un indice Ki-67 inferiore all’1% e positività per melan-A, e sono lesioni benigne 4). Raramente si associano a fragilità della zonula, e durante l’intervento di cataratta si può osservare una diastasi zonulare diffusa 4).
L’immunoistochimica della membrana di Descemet rimossa da occhi con sindrome ICE mostra un pattern di espressione mista di marcatori epiteliali (AE1/AE3, CK8/18) e marcatori endoteliali (CD56, vimentina), confermando che la metaplasia epiteliale è la patologia essenziale della sindrome ICE7). È importante la diagnosi differenziale con la down-growth epiteliale (crescita epiteliale interna), ma si decide in base alla combinazione del decorso clinico e dei reperti istologici.
QCome si differenzia la sindrome ICE dalla distrofia corneale polimorfa posteriore (PPMD)?
A
Il punto di differenziazione più semplice è che la sindrome ICE è sporadica e unilaterale, mentre la PPMD è autosomica dominante e bilaterale. Anche la microscopia speculare è diversa: nella sindrome ICE mostra aree scure con un punto luminoso centrale (cellule ICE), mentre la PPMD presenta tipiche vescicole o strutture a banda. La PPMD può presentare sinechie anteriori dell’iride o edema corneale, ma non ha differenze di genere ed è congenita, quindi il decorso clinico è diverso.
Pinheiro-Costa J, Maia J, Branco A, et al. Two-step iridocorneal endothelial syndrome management: endocapsular intraocular lens implantation and Descemet’s stripping automated endothelial keratoplasty. Case Rep Ophthalmol. 2023;14(1):478-486.
Guler Canozer D, Unlu M, Gultekin Irez B, Ozkurt Y. In vivo confocal microscopy and anterior segment optical coherence tomography findings in iridocorneal endothelial syndrome. Turk J Ophthalmol. 2024;54(5):325-330.
Bouvarel H, Hamard P, Agard E, Billant J, El Chehab H, Dot C. Macular edema in Cogan-Reese syndrome. American journal of ophthalmology case reports. 2022;25:101318. doi:10.1016/j.ajoc.2022.101318. PMID:35128161; PMCID:PMC8810354.
Chhadva P, Del Valle Estopinal M, Farid M. Iris Nevus (Cogan-Reese) Syndrome Presenting with Zonular Dehiscence during Cataract Extraction. Case reports in ophthalmology. 2022;13(2):435-440. doi:10.1159/000524823. PMID:35950024; PMCID:PMC9247537.
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.
Saleki M, Lee P, Thaung C, Ashena Z. Descemet’s membrane endothelial keratoplasty in an eye with iridocorneal endothelial syndrome and rare association of corneal ectasia. Ther Adv Ophthalmol. 2025;17:25158414251343968.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.