Iris malignant melanoma is a malignant tumor originating from melanocytes of the uveal tissue (iris, ciliary body, choroid), specifically arising in the iris. It accounts for only about 2% of all uveal melanomas, making it the rarest site compared to choroidal (over 90%) and ciliary body (about 7%) origins.
The incidence is 0.025 per 100,000 population (about 1/20 of that in Western countries), and iris cases are only a subset. It is more common in Caucasians and people with light-colored irises, and rare in East Asians. Compared to choroidal and ciliary body melanomas, it tends to have lower malignancy and slower progression.
Iris malignant melanoma may also result from invasion from the ciliary body. Large size, growth tendency, irregular shape, and angle invasion are indicators of malignancy. Small tumors may be amenable to local resection (iridectomy).
Metastasis is exclusively hematogenous (since the uvea lacks lymphatics), and when metastasis occurs, the liver is the most common site 2). While the 12-year mortality rate for choroidal melanoma is about 40%, iris melanoma has a much lower metastasis rate and a better prognosis.
Iris malignant melanoma tends to occur at a younger age, and can be found in age groups earlier than the peak incidence of choroidal melanoma (around 60 years) 1).
QHow rare is iris melanoma?
A
Even among uveal melanomas overall, it is a rare cancer with an incidence of 2 to 8 per million in Caucasians, and iris cases account for only about 2%. In Japanese, the incidence of all uveal melanomas is 0.025 per 100,000 (about 1/20 of Western countries), and iris cases are even rarer. If a pigmented lesion is found in the iris, examination at a specialized facility is important.
Trobe JD. The Eyes Have It. Kellogg Eye Center, University of Michigan. 2011. Figure 1. Source ID: commons.wikimedia.org/wiki/File:Iris_melanoma.jpg. License: CC BY 3.0.
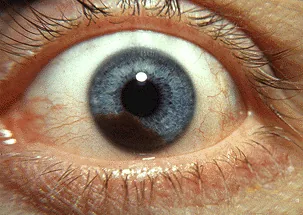
Clinical photograph of iris malignant melanoma; a pigmented solid tumor protrudes from the iris surface, accompanied by pupillary distortion. Corresponds to the solid pigmented iris tumor and pupillary distortion discussed in the section “2. Main symptoms and clinical findings”.
Early small lesions are often asymptomatic and may be discovered incidentally during health checkups or examinations for other diseases. As the lesion grows, the following symptoms appear.
Decreased vision/blurring: due to enlargement or complications.
Photophobia/eye pain: may be caused by secondary glaucoma due to angle invasion or elevated intraocular pressure.
Change in iris color: may be noticed as enlargement or color change of a unilateral iris pigmented spot.
Lens displacement: in the ciliary body infiltration type, displacement of the lens-iris diaphragm may occur.
It appears as a solid, pigmented to non-pigmented tumor on the iris surface. Clinical indicators suggesting malignancy are as follows:
Large size (large diameter)
Growth tendency (enlargement on follow-up)
Irregular shape
Angle invasion/elevated intraocular pressure
Rubeosis iridis (ciliary body infiltration type)
Lens subluxation (ciliary body infiltration type)
Nodular type
Morphology: Well-defined, dome-shaped elevated mass
Metastatic risk: Metastasis is rare. This is the type with the best prognosis1)
Features: The mass is confined to the iris stroma with minimal invasion of the surrounding iris
Treatment indication: Small lesions may be treated with iridectomy
Diffuse type
Morphology: Flat, diffuse infiltrative lesion involving the entire iris. Borders are indistinct
Metastatic risk: More likely to metastasize than the nodular type1)
Features: Tends to cause elevated intraocular pressure and angle infiltration. May be discovered as iris heterochromia (difference in iris color between the two eyes)
Treatment indication: Local excision is difficult, and radiation therapy or enucleation is often required
The onset is mainly sporadic, and the exact cause is unknown. The main risk factors are listed below.
Uveal nevus: The most common risk factor. About 10% arise from a pre-existing nevus1).
Light-colored iris, fair skin, tendency to sunburn: More common in Caucasians and Northern Europeans.
Congenital ocular melanocytosis: A rare risk factor.
Ultraviolet radiation (UVR): UVR-induced signatures have been reported in some iris melanomas 3). Similarities to cutaneous melanoma suggest a role for UV radiation. The mechanism of development may differ from that of choroidal melanoma.
Family history: Germline mutations in the BAP1 gene (BAP1 tumor predisposition syndrome) increase the risk of uveal melanoma 1).
The genetic mutation profile of uveal melanoma (including iris melanoma) is as follows.
Mutated Gene
Mutation Frequency
Metastasis Risk / Features
GNAQ/GNA11
83–89%
Mutually exclusive initiating mutations. No direct association with metastasis risk.
BAP1
Approximately 45%
Highest metastasis risk (peak at 3.5 years), class 2 tumors
SF3B1
Approximately 23%
Intermediate risk. Characterized by late metastasis (peak at 7 years).
EIF1AX
Approximately 17%
Lowest metastasis risk
GNAQ/GNA11 mutations are initiating mutations that impair GTPase activity and lead to constitutive activation, but they are not directly associated with tumor size or metastasis risk. Secondary driver mutations (BAP1, SF3B1, EIF1AX) occur almost completely mutually exclusive of each other and are used for metastasis risk stratification.
BAP1 mutations are associated with the highest metastasis risk, while EIF1AX mutations indicate the lowest metastasis risk. This mutation information can be obtained via fine-needle aspiration biopsy and is used to personalize metastasis surveillance plans 1).
QHow do genetic mutations in iris melanoma relate to prognosis?
A
GNAQ/GNA11 mutations are present in approximately 85% of cases but are not directly associated with metastasis risk. The risk of metastasis is determined by secondary mutations: BAP1 mutations (highest risk, metastasis peak at 3.5 years), SF3B1 mutations (moderate risk, late metastasis peak at 7 years), and EIF1AX mutations (lowest risk). Obtaining these genetic profiles through biopsy allows for personalized surveillance frequency.
Diagnosis is primarily based on a comprehensive evaluation of imaging tests and clinical findings. For iris melanoma, examinations focusing on the anterior segment are important.
Slit-lamp microscopy: Basic for detailed observation of the anterior segment. Evaluates tumor morphology, neovascularization, and angle invasion. Gonioscopy confirms tumor invasion into the angle.
UBM (Ultrasound Biomicroscopy): Essential for evaluating ciliary body invasion. Used to monitor tumor size and for follow-up of small cases. The presence of ciliary body invasion directly affects treatment strategy.
Anterior segment OCT: Evaluates the surface morphology of iris tumors. Used adjunctively to assess internal tumor structure.
MRI: Melanin’s paramagnetic properties cause high signal on T1-weighted images and low signal on T2-weighted images. Useful for evaluating extraocular extension and ciliary body involvement. However, this finding is non-specific.
FDG-PET/CT: Used for metastasis detection. Reports suggest it has superior sensitivity and specificity compared to CT alone 2).
Fine-needle aspiration biopsy is performed depending on the situation. Increasingly, the goal is to obtain genetic profiles (GNAQ/GNA11, BAP1, SF3B1, EIF1AX). The indication is carefully considered due to the risk of tumor seeding.
Metastatic iris tumor: Metastasis from breast or lung cancer. Often white to milky white and multiple. History of systemic malignancy and bilateral involvement are clues for differentiation.
Cogan-Reese syndrome (iris nevus syndrome): A type of iridocorneal endothelial syndrome. Presents with iris nodules, corneal endothelial abnormalities, and elevated intraocular pressure.
Iris melanocytoma: Benign black mass. Histologically benign, but malignant transformation has been reported.
QIf the color of the iris changes, what tests should be done?
A
First, examine the anterior segment in detail with a slit-lamp microscope to assess the characteristics, extent, and angle invasion of the mass. Next, use UBM to check for ciliary body invasion and anterior segment OCT to record the surface morphology. If malignancy is suspected, evaluate extraocular extension with MRI and perform liver ultrasound and whole-body CT for metastasis screening. Referral to a specialized facility (ocular oncology specialist) is recommended.
For small lesions that are difficult to distinguish from iris nevus, tumor size is regularly monitored using UBM or anterior segment OCT. Treatment is initiated when signs of growth or malignancy (enlargement, irregular shape, angle invasion, increased intraocular pressure) appear.
First choice for small iris melanoma. The iris containing the tumor is resected (iridectomy). If the ciliary body is also involved, iridocyclectomy is performed. This allows eye preservation and enables pathological diagnosis and genetic profiling from the resected specimen.
Indicated for medium-sized tumors (when local resection is difficult).
Brachytherapy: I-125 (iodine-125) or Ru-106 (ruthenium-106) plaque is sutured onto the sclera over the tumor. A prescribed dose of approximately 90 Gy is delivered to the tumor apex.
Heavy particle therapy (proton beam, carbon ion): Concentrates dose on the tumor using the Bragg peak effect, minimizing dose to surrounding normal tissue.
CyberKnife (stereotactic radiotherapy): A non-invasive option.
Indicated for large tumors or when eye preservation is difficult (e.g., diffuse type, extensive ciliary body invasion). It remains an important option today.
For metastatic uveal melanoma, tebentafusp is recommended for HLA-A*02:01-positive patients1,5).
In a phase III randomized controlled trial by Nathan et al. (2021), tebentafusp showed a significant improvement in overall survival compared to investigator’s choice (mainly pembrolizumab) in untreated metastatic uveal melanoma patients (HLA-A*02:01 positive) (mOS: 21.7 months vs 16.0 months) 4).
Tebentafusp is a T-cell receptor bispecific fusion protein (ImmTAC) that recognizes the tumor-associated antigen gp100 on HLA-A02:01 complexes and activates T cells. Administration is performed weekly via intravenous injection after confirmation of HLA-A02:01 positivity (escalation: 20 mg → 30 mg → 68 mg).
Ocular adverse effects such as choroidal thinning, fundus depigmentation, and cutaneous depigmentation have been reported during administration, requiring regular ophthalmic monitoring 7). Since gp100 is also expressed on normal choroidal melanocytes, these side effects are explained by the mechanism 7).
Post-treatment monitoring of the affected eye is performed regularly with slit-lamp microscopy, UBM, and anterior segment OCT. Local recurrence checks and metastasis surveillance are conducted concurrently. Liver metastasis surveillance every 6 months for 5 years, then annually for 10 years after treatment is recommended 1).
Iris melanoma develops from neoplastic proliferation of iris melanocytes. The pathogenesis of uveal melanoma differs from that of cutaneous melanoma and involves distinct molecular pathways.
Mutations at Q209 of GNAQ/GNA11 are the most common initiating mutations, impairing GTPase activity and leading to constitutive GTP-bound activation. This results in sustained activation of multiple signaling pathways, including the MAPK pathway (Ras/RAF/MEK/ERK) 5).
Secondary driver mutations (BAP1, SF3B1, EIF1AX) occur almost completely mutually exclusive of each other. BAP1 mutations are classified as class 2 (high metastatic risk) and are strongly associated with monosomy 3. Patients with SF3B1 mutations have a relatively favorable median overall survival after metastasis, characterized by late-onset metastasis.
Iris melanoma tends to metastasize less frequently than choroidal and ciliary body melanoma. Factors associated with this characteristic include the following:
The iris has abundant blood flow, but tumor progression takes time.
Nodular type rarely metastasizes, while diffuse type is more likely to metastasize 1).
In iris melanoma, UVR-induced mutation signatures have been identified in some cases (Johansson et al. 2020) 3). This is thought to be related to the anatomical characteristic that the iris is located in the anterior segment and is more exposed to ultraviolet light than the choroid. This finding suggests that iris melanoma may have a pathogenesis similar to cutaneous melanoma 3).
Since there are no lymphatic vessels in the uvea, all metastases occur hematogenously 2). Circulating tumor cells travel through the bloodstream and cause liver metastases due to strong liver tropism (seed and soil theory). Metastases may appear more than 25 years after treatment of the primary tumor 2), which is why long-term surveillance is necessary.
Intratumoral heterogeneity exists, and different morphological regions may have different genetic profiles 6), so the location of biopsy sampling may affect the accuracy of prognosis prediction.
7. Latest Research and Future Perspectives (Investigational Reports)
Tebentafusp is the first treatment for metastatic uveal melanoma to show a significant improvement in overall survival in a phase III trial 4). Three-year follow-up data reported in 2023 (Hassel et al.) confirmed sustained survival improvement, with a 3-year overall survival rate of 27% (vs. 9% in the control group) 9).
Since gp100 is also expressed in iris melanoma, the drug may be considered for metastatic cases arising from iris melanoma 1). Ocular side effects during administration (choroidal thinning, fundus depigmentation) may be irreversible, and collaboration with ophthalmology specialists is necessary 7).
Combination of a Gαq inhibitor (YM-254890) and a MEK inhibitor (trametinib/binimetinib) has shown synergistic antitumor effects in vitro and in vivo 5). While MAPK signaling recovers within 24 hours with Gαq inhibition alone, the combination with a MEK inhibitor suppresses this recovery 5). Evaluation in clinical trials is ongoing.
In uveal melanoma, the efficacy of immune checkpoint inhibitors is significantly more limited compared to cutaneous melanoma 7), with response rates of approximately 5% for monotherapy and 12–18% for combination therapy 1). This is thought to be due to the low tumor mutational burden (TMB) and characteristics of the immune microenvironment in uveal melanoma 2).
However, cases with germline or somatic loss-of-function mutations in the MBD4 (methyl-CpG binding domain-4) gene have been reported to potentially have increased sensitivity to checkpoint inhibitors 2).
Johansson et al. (2020) identified a UVR-induced mutational signature in iris tumors through whole-genome analysis 3). This is the first report indicating that iris melanoma, among uveal melanomas, harbors ultraviolet-related genetic alterations, providing important insights into differences in pathogenesis from choroidal melanoma. This finding may have implications for the role of UV protection and future prevention and treatment strategies.
9-ARG prognostic signature and immune microenvironment
A 9-autophagy-related gene signature (9-ARG) has been shown to be useful for prognosis prediction in uveal melanoma 8). In the high-risk group, the IL6-JAK-STAT3 pathway and angiogenesis-related pathways are enriched, and although immune cell infiltration (CD8 T cells, activated memory CD4 T cells) is increased, it presents an immunosuppressive phenotype, paradoxically associated with poor prognosis 8). This is thought to be related to the eye being an immune-privileged organ.
Carter TJ, Broadfoot J, Coupland SE, et al. Uveal Melanoma UK national guidelines: 2023 update. Eur J Cancer. 2023;185:61-73.
Rantala ES, Hernberg MM, Piperno-Neumann S, Grossniklaus HE, Kivelä TT.. Metastatic uveal melanoma: The final frontier. Prog Retin Eye Res. 2022;90:101041. doi:10.1016/j.preteyeres.2022.101041. PMID:34999237.
Johansson PA, Brooks K, Newell F, Palmer JM, Wilmott JS, Pritchard AL, Broit N, Wood S, Carlino MS, Leonard C, Koufariotis LT, Nathan V, Beasley AB, Howlie M, Dawson R, Rizos H, Schmidt CW, Long GV, Hamilton H, Kiilgaard JF, Isaacs T, Gray ES, Rolfe OJ, Park JJ, Stark A, Mann GJ, Scolyer RA, Pearson JV, van Baren N, Waddell N, Wadt KW, McGrath LA, Warrier SK, Glasson W, Hayward NK.. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat Commun. 2020;11(1):2408. doi:10.1038/s41467-020-16276-8. PMID:32415113; PMCID:PMC7229209.
Nathan P, Hassel JC, Rutkowski P, et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med. 2021;385(13):1196-1206. doi:10.1056/nejmoa2103485.
Sriramareddy SN, Smalley KSM. MEK-ing the most of it: strategies to co-target Gαq and MAPK in uveal melanoma. Clin Cancer Res. 2021;27(5):1217-1219. doi:10.1158/1078-0432.CCR-20-4530. PMID:33355300; PMCID:PMC7925419.
Cristina Fonseca, Rita Pinto-Proença, Sabrina Bergeron, Luís Miguel Pires, Júlia Fernandes, Isabel Marques Carreira, et al. Intratumoral Heterogeneity in Uveal Melanoma. Ocul Oncol Pathol. 2020;7(1):17-25. doi:10.1159/000508517.
Krohn J, Vinnem LIH, Jansson RW, Straume O. Fundus hypopigmentation and choroidal thinning associated with tebentafusp therapy: report of a case and literature review. BMC ophthalmology. 2025;25(1):464. doi:10.1186/s12886-025-04274-7. PMID:40817046; PMCID:PMC12357441.
Chuah S, Chew V. Immune implication of an autophagy-related prognostic signature in uveal melanoma. Bioscience reports. 2021;41(8). doi:10.1042/BSR20211098. PMID:34374416; PMCID:PMC8380919.
Jessica C. Hassel, Sophie Piperno-Neumann, Piotr Rutkowski, Jean-Francois Baurain, Max Schlaak, Marcus O. Butler, Ryan J. Sullivan, Reinhard Dummer, et al. Three-Year Overall Survival with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med. 2023;389(24):2256-2266. doi:10.1056/nejmoa2304753.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.