Uveal melanoma (UM) is a malignant tumor arising from melanocytes of the uvea (iris, ciliary body, and choroid). It is the most common primary intraocular tumor in adults, with over 90% originating in the choroid, about 7% in the ciliary body, and 2% in the iris. This article focuses on choroidal and ciliary body melanoma.
The incidence is about 1/20 of that in Western countries, with 0.025 per 100,000 population. In Caucasians, the incidence is 2 to 8 per million, with a slight male predominance and a peak onset around age 60. Less than 1% of all uveal melanomas occur in individuals under 18 years old. Worldwide, approximately 6 million people are diagnosed with ocular melanoma annually6).
The mode of metastasis is exclusively hematogenous (since the uvea lacks lymphatics), with a strong predilection for the liver. Liver metastases are found in 70–90% of cases, with other sites including the lungs, bones, and skin6). More than 62% of metastases become clinically apparent within 5 years after treatment of the primary tumor, but the remainder may be detected more than 25 years later. Because metastases may become evident several years to more than a decade after treatment, long-term monitoring for metastasis is necessary.
The median overall survival (OS) after metastasis is estimated at 10–13 months based on a meta-analysis (2,494 patients, 78 papers), with approximately 2% surviving beyond 5 years and 13% surviving beyond 3 years 13). One-year survival rates are reported as 43–52%. The 12-year mortality rate is approximately 40%, nearly identical regardless of the choice of local treatment 2). The 5-year survival rate for medium-sized tumors is 70–80% (no difference between eye-preserving therapy and enucleation), and it is clear that there is no significant difference in prognosis based on treatment modality.
For intermediate- to high-risk patients, it is recommended to continue liver imaging every 6 to 12 months for 10 years14).
QHow rare is choroidal melanoma?
A
In Europe and the United States, the incidence is 2 to 8 per million among Caucasians, making it the most common primary intraocular tumor in adults, but the absolute number is small. The frequency of occurrence is about 1/20 of that in Europe and the United States, at 0.025 per 100,000 population. It is more common in Caucasians, and even rarer in Asians.
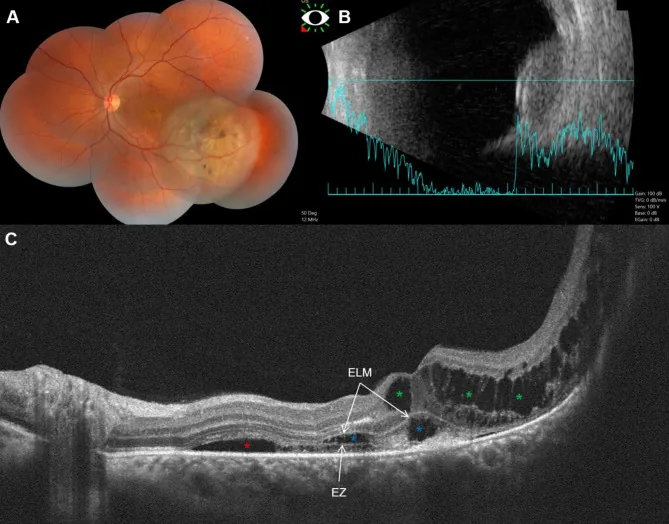
Fouad YA, et al. Bacillary layer detachment with malignant choroidal tumors: a case series. BMC Ophthalmol. 2023. Figure 1. PMCID: PMC10077734. License: CC BY.
Panel (A) shows a non-pigmented elevated lesion and pigmentation in the left eye, (B) shows a dome-shaped tumor with low to moderate internal reflectivity, and (C) shows subretinal fluid, cystoid edema, and basal laminar deposition (BALAD). This corresponds to choroidal melanoma, which is discussed in the section “2. Main symptoms and clinical findings.”
Approximately 30% of patients are asymptomatic at the initial visit, and the condition is discovered incidentally during health checkups or examinations for other diseases. When symptoms appear, the breakdown is as follows: decreased vision 38%, photopsia 9%, floaters 7%, peripheral visual field defect 6%, and eye pain 2%.
While the tumor is small and located in the peripheral fundus, it is generally asymptomatic. As it grows, the following symptoms appear.
Photopsia and floaters: These are observed from a relatively early stage.
Visual field defect: As the tumor enlarges, it spreads from the periphery toward the center.
Metamorphopsia and decreased visual acuity: These become prominent when the macula is involved. Serous retinal detachment often occurs concurrently, making visual field and visual acuity deficits apparent.
Vitreous hemorrhage: Sudden vision loss may occur due to bleeding from the tumor.
Eye pain: Extremely rare, occurring in less than 2% of choroidal melanoma cases. The main causes are secondary glaucoma due to elevated intraocular pressure or tumor necrosis4).
Symptoms of ciliary body melanoma exhibit the following characteristics due to the specificity of its anatomical location5).
Painless vision loss (blurring): The most common complaint.
Astigmatism due to lens displacement: Forward movement of the lens and iris diaphragm caused by the tumor.
Painless visual field defect when the visual axis is involved.
Painful vision loss due to rapid intraocular pressure elevation: Caused by secondary angle-closure glaucoma.
Secondary intraocular pressure elevation is observed in up to 17% of cases at the time of ciliary body melanoma diagnosis3).
Pigmentation: 55% are heavily pigmented, 30% mixed, and 15% amelanotic, showing a variety of colors.
Mushroom-shaped configuration: Approximately 20% break through Bruch’s membrane and take on a mushroom (button) shape. They rapidly enlarge after penetrating Bruch’s membrane.
Subretinal fluid: Frequently accompanied by serous retinal detachment.
Ciliary Body Melanoma
Size at discovery: Due to anatomically hidden locations, they are often relatively large when found.
Sentinel vessel: Dilated and tortuous episcleral vessels directly over the tumor are frequently observed.
Anterior segment changes: Anterior displacement of the lens-iris diaphragm, often leading to secondary angle-closure glaucoma.
Extraocular extension: There is a risk of extraocular extension via the emissary canal.
Ultrasound Hollow: internal hypoechogenicity on ultrasound
Halo absent: absence of a halo (light ring)
Drusen absent: absence of drusen
When there are zero risk factors, the probability of growth within five years is only 3%, but with one risk factor it rises to 38%, and with two or more it exceeds 50%.
QHow can we differentiate between a choroidal nevus (mole) and melanoma?
A
Evaluation is performed using the eight risk factors of TFSOM-UHHD. With zero risk factors, the probability of growth within five years is only 3%, but with two or more factors, it rises to over 50%. Regular follow-up with ultrasound and fundus photography is necessary.
Onset is mainly sporadic. The cause is unknown, but abnormalities in tumor suppressor genes and oncogenes, as well as sun exposure, are thought to be involved. The main risk factors are shown below.
Light iris color, fair skin, sunburn-prone constitution: Common among Caucasians and Northern Europeans.
Uveal nevus: The most common risk factor. Approximately 10% arise from a pre-existing nevus. About 6% of Caucasians have a choroidal nevus, with a malignant transformation rate of 1/5000 to 1/8845.
Family history of uveal melanoma: Rare but reported.
Ultraviolet light: Its role is considered uncertain.
The correspondence between gene mutations and metastasis risk is shown below.
Mutant gene
Mutation frequency
Metastasis risk/characteristics
GNAQ/GNA11
83–89%
Mutually exclusive initiating mutations. No direct association with metastatic risk.
BAP1
45%
Highest metastasis risk (large peak at 3.5 years), class 2
SF3B1
23%
Moderate risk. Characterized by late metastasis (large peak at 7 years).
EIF1AX
17%
Lowest risk of metastasis
GNAQ/GNA11 mutations are considered early events in tumor formation, and these mutations alone are not significantly associated with tumor size or risk of metastasis. Intratumoral heterogeneity exists, and genetic profiles may differ between morphologically distinct regions (while monosomy 3 is common across regions, 6q deletion may be limited to pigmented areas in some cases) 2).
BAP1 mutations are associated with the highest risk of metastasis1), while EIF1AX mutations are associated with the lowest1).
Histologically, it is classified into spindle cell type and epithelioid cell type. Cases with a higher proportion of epithelioid cells are considered to have a poorer prognosis. Mixed types also exist.
QHow do genetic mutations in uveal melanoma relate to prognosis?
A
BAP1 mutations carry the highest risk of metastasis (peak at 3.5 years), SF3B1 mutations are characterized by late metastasis (peak at 7 years), and EIF1AX mutations show the lowest risk. Additionally, detection of chromosome 3 deletion (monosomy 3) is associated with a high rate of metastasis and poor prognosis. These mutation data are obtained through fine-needle aspiration biopsy and are used to individualize metastasis surveillance plans.
Serial fundus photography is extremely important for documenting tumor growth, and the use of wide-angle fundus imaging (e.g., Optos) is also useful. In fundus autofluorescence, the autofluorescent properties of lipofuscin are utilized to identify bright orange fluorescence (orange pigment) that is brighter than drusen.
FA: In the early to mid-phase of angiography, intratumoral vessels and multiple punctate or patchy hyperfluorescence are observed, with diffuse hyperfluorescence and dye leakage in the late phase. Blocking fluorescence (hypofluorescence) due to pigment and hyperfluorescence due to lipofuscin deposition at the RPE level are mixed.
ICGA: Tumor blood vessels are visualized more clearly (dual circulation pattern). Excellent for assessing intratumoral vasculature.
It shows high signal on T1-weighted images and low signal on T2-weighted images. It is also used to track tumor size after I-125 plaque irradiation 7).
In SPECT using 123I-IMP (iodoamphetamine) as a tracer, abnormal accumulation consistent with the affected eye is observed 24 hours after intravenous injection, making it an excellent test with high sensitivity and specificity. FDG-PET may also be used for diagnosis.
Liver ultrasound: Used for screening and surveillance. Metastases are generally hypoechoic (67%). Sensitivity is high at 95–100%, and due to the lack of radiation exposure, it is suitable for regular surveillance13).
Liver MRI: Higher ability to identify liver metastases than CT. Sensitivity 83–100% with no radiation exposure13). Recommended as a non-ionizing radiation imaging test14).
CT: Essential for staging lung and liver metastases. Although it involves radiation exposure, it is mandatory for initial staging evaluation 14).
Molecular testing of tumor tissue or cells obtained by needle biopsy is important for stratifying the risk of metastasis.
Monosomy 3 (loss of chromosome 3): When detected, it is associated with a high rate of metastasis and poor prognosis. The 5-year survival rate decreases from approximately 100% to about 50%14).
Chromosome 8q gain and 1p loss: Correlated with decreased survival rate 14).
Immunohistochemistry for BAP1 nuclear expression: Important for prognostic stratification. Loss of BAP1 nuclear expression suggests BAP1 mutation and is associated with high metastatic risk 14).
Performed in cases where the clinical diagnosis is uncertain. It is usually performed concurrently with plaque brachytherapy insertion. Although there is debate due to the risk of tumor seeding, it is increasingly performed for prognostic stratification through gene expression profiling. Considering intratumoral heterogeneity, sampling from multiple morphologically distinct areas is desirable 2).
S-100, HMB-45, MART-1 (MelanA), and vimentin are positive. For confirmation, it is recommended to use at least two melanocytic markers in combination with cytokeratin (epithelial marker). Semi-quantitative assessment of the proliferation index using Ki-67 is also performed. In ciliary body melanoma, strong positivity for HMB-45 has been confirmed 5). BAP1 nuclear staining plays an important auxiliary role in prognosis classification 2).
The goals of treatment are threefold: (1) maintaining useful vision in the affected eye, (2) destroying the tumor, and (3) preventing metastasis and recurrence.
While differentiation from choroidal nevus remains unclear, strict follow-up is continued with fundus photography and B-mode ultrasonography. For choroidal nevus (thickness less than 2 mm, asymptomatic), re-examination is performed 3 months after the initial examination, followed by follow-up every 6 months. For lesions less than 3 mm thick, fundus photography, FA, and A/B-mode ultrasonography are performed initially, re-examined after 3 to 4 months, and then fundus photography is continued every 6 to 12 months for life. Small tumors with three or more risk factors should be treated promptly without waiting for growth records.
106Ru plaque brachytherapy: A plaque containing ruthenium-106 (beta emitter) is sutured onto the sclera over the tumor. Due to beta radiation, the intra-tissue irradiation distance is short, making it suitable for small to medium-sized tumors. Facilities capable of performing this procedure are limited.
I-125 plaque: A COMS plaque containing iodine-125 is sutured onto the sclera over the tumor site. The prescribed radiation dose to the tumor apex is 90 Gy. This method is primarily used in North America.
Indications: First-line treatment for small to medium-sized tumors.
Recurrence: 80% of local recurrences occur within 3 years. 98% are detectable on fundus color photography.
Visual prognosis: According to COMS 3-year data, 43–49% of patients have visual acuity of 20/200 (equivalent to 0.1) or worse. Radiation retinopathy is the main cause10).
Proton beam therapy
Characteristics: Particle beam therapy that concentrates radiation dose on the tumor using the Bragg peak effect. Eye-preserving therapy.
Indications: Eye-preserving option for small to medium-sized tumors.
Treatment efficacy: Cases have been reported in which complete tumor regression was achieved 6 months after proton beam therapy.
Advantages: Low radiation dose to surrounding normal tissues.
The indications for external beam radiation therapy, such as heavy particle therapy and CyberKnife, are expanding. While eye preservation is possible, visual function is often lost due to complications such as optic neuropathy and neovascular glaucoma. Although the number of facilities where it can be performed is limited, it is positioned as one of the treatment options.
Transpupillary thermotherapy (TTT) is indicated for small choroidal melanoma (thickness ≤3 mm). The main parameters are shown below9).
Parameter
Setting value
Wavelength
810 nm (near-infrared)
Output
400–1,000 mW
Spot diameter
3,000 μm
Irradiation time
1–3 minutes per spot
Depth
3–4 mm
In “sandwich therapy,” which combines transpupillary thermotherapy and brachytherapy (plaque), the 5-year recurrence rate has been reported as 3% with 125I and 10% with 106Ru9).
If the tumor is relatively small and especially located anteriorly, local resection may be performed to remove only a portion of the sclera and the tumor.
For large tumors (COMS large classification: apical height >10 mm, largest basal diameter >16 mm) where eye preservation is difficult, enucleation is indicated. It remains one of the treatment options today. For orbital recurrence after enucleation, surgical and radiation therapy should be considered in a multidisciplinary conference. Radiation therapy is recommended at 45–50 Gy in 20 fractions, with >2 Gy per fraction.
Systemic treatment for metastatic uveal melanoma has significantly fewer options compared to cutaneous melanoma.
Tebentafusp: A T-cell receptor bispecific fusion protein for HLA-A*02:01-positive patients. Phase III trial demonstrated significant OS improvement (median 6-month extension compared to investigator’s choice control), making it the first effective drug for metastatic uveal melanoma14). Dosing protocol: weekly IV (escalation from 20 to 30 to 68 mg)7).
Immune checkpoint inhibitors: The response rate as monotherapy is approximately 5%, and 12–18% with nivolumab plus ipilimumab. The effect is limited compared to cutaneous melanoma14).
MEK inhibitors: Low activity rates as monotherapy or in combination14).
Liver-directed therapy: Options include liver resection (when R0 is possible), percutaneous hepatic perfusion (PHP), selective internal radiation therapy (SIRT), and transarterial chemoembolization (TACE). PHP and SIRT show the longest overall survival14).
Median OS after metastasis: 10–13 months in meta-analysis, 13% survive >3 years13).
Post-treatment monitoring of the affected eye is performed every 6 months for 2–5 years, then annually thereafter. Cases with complete tumor regression can be transitioned to a local optometrist, and cases with enucleation plus R0 resection can be transitioned to an ocularist after wound healing.
For metastasis surveillance, liver MRI or ultrasound every 6 months is recommended for high-risk patients, and liver imaging every 12 months for low-risk patients 14). Non-ionizing radiation imaging (MRI, ultrasound) is recommended to avoid radiation exposure. For high-risk patients with monosomy 3, metastasis monitoring should be intensified.
QAre there treatments that can preserve the eye?
A
For small to medium-sized tumors, options include brachytherapy (such as 106Ru plaque therapy), proton beam therapy, heavy particle therapy, and CyberKnife, and eye preservation is often possible. For large tumors, enucleation may be necessary, but it has been shown that the choice of treatment does not affect metastasis rate or overall survival. Therefore, while prioritizing life prognosis, the feasibility of preserving visual function is considered.
QWhat are the treatment options if metastasis is found?
A
If HLA-A*02:01 positive, tebentafusp becomes the first-line candidate. It is the first drug shown to significantly improve overall survival in a phase III trial. For liver-limited metastases, consider liver resection if R0 resection is possible. Liver-directed therapies (PHP, SIRT, TACE, etc.) are also options. The efficacy of immune checkpoint inhibitors is limited compared to cutaneous melanoma (monotherapy response rate approximately 5%), and it is important to conduct a multidisciplinary conference at a specialized facility before starting treatment.
The pathogenesis of uveal melanoma involves a unique molecular pathway distinct from that of cutaneous melanoma.
The Q209 mutation in GNAQ/GNA11 is the most common, and R183 and G48L mutations have also been identified1). These mutations impair GTPase activity, leading to a constitutively active GTP-bound state1). GNAQ/GNA11 mutations persistently activate multiple signaling pathways, including the MAPK pathway (Ras/RAF/MEK/ERK)1). GNAQ/GNA11 mutations are early events in tumorigenesis and are not significantly associated with tumor size or metastatic risk.
Secondary driver mutations (BAP1, SF3B1, EIF1AX) occur almost completely mutually exclusively and are important for stratifying metastasis risk. BAP1 mutations are classified as class 2 (high metastasis risk), and the median post-metastasis overall survival for SF3B1 mutation carriers is 3.9 years (95% CI 2.3–6.2), with a 12-month overall survival rate of 94%, indicating a relatively favorable course. In metastatic lesions, GNAQ (57%) and GNA11 (36%) are also detected mutually exclusively.
As rare initiating mutations, CYSLTR2 and PLCB4 mutations are detected in almost all remaining uveal melanomas.
Since the uvea lacks lymphatic vessels, all metastases occur via the bloodstream 6). In the early stage, the lesion is small and flat, but as it becomes elevated, it breaks through Bruch’s membrane and grows rapidly. Serous retinal detachment may occur around the lesion.
Circulating tumor cells are detected in 10–88% of patients. The notable tropism for the liver is explained by the seed and soil theory. Micrometastases can occur early, even during the asymptomatic phase of the primary tumor. Monosomy 3 is found in 70–100% of metastases, and BAP1 mutations in 60–80%. Cases of metastasis to rare sites such as the thyroid have also been reported11).
Two growth patterns of liver metastasis are known: sinusoidal infiltrative and periportal nodular. In the infiltrative type, collagen production leads to pseudosinusoid formation, while in the nodular type, VEGF-induced angiogenesis is the main mechanism13).
Intratumoral VEGF concentration is significantly higher than in healthy eyes and positively correlates with tumor basal diameter and height 3). Systemic administration of anti-VEGF drugs (bevacizumab) has shown metastasis-suppressing effects in mouse models, but contradictory results have been reported where intravitreal administration accelerated tumor growth 3).
A case of rapid enlargement of ciliary body melanoma after intravitreal bevacizumab injection (basal diameter from 2.51 to 18.0 mm, height from 6.23 to 11.0 mm over 7 weeks) has been reported3). The median doubling time for typical choroidal melanoma is 154–511 days, making this rapid growth unusual.
Intratumoral heterogeneity and immune microenvironment
Intratumoral heterogeneity exists in both morphological and genetic aspects, affecting the accuracy of biopsy-based prognosis prediction 2). This is why sampling from multiple sites is recommended 2).
An autophagy-related 9-gene signature (9-ARG: IKBKE, BNIP1, ITGA6, FKBP1A, DLC1, PRKCD, GABARAPL1, LMCD1, TUSC1) has been shown to be useful for predicting prognosis in uveal melanoma (validated in 80 TCGA cases + 150 GEO cases)8). In the high-risk group, the IL6-JAK-STAT3 pathway, angiogenesis, and reactive oxygen species pathways are enriched, and immune cell infiltration (CD8 T cells, activated memory CD4 T cells) is increased, but a paradoxical finding has been reported that this is associated with an immunosuppressive phenotype and poor prognosis8). This is thought to be related to the eye being an immune-privileged organ.
7. Latest Research and Future Prospects (Reports at the Research Stage)
Tebentafusp is a T-cell receptor bispecific fusion protein designed for HLA-A02:01-positive patients 7). It recognizes the tumor-associated antigen gp100 on the HLA-A02:01 complex and exerts antitumor effects by activating T cells.
In a phase III trial, patients with HLA-A*02:01-positive metastatic uveal melanoma showed a significant improvement in overall survival (median extension of 6 months compared with the investigator’s choice control) 14).
In a case report by Krohn et al. (2025), a patient with metastatic uveal melanoma who received tebentafusp (weekly IV escalation: 20→30→68 mg) based on a phase III trial showed a favorable course with stable liver metastases and no new lesions after 26 months of treatment7). In this patient, the right eye central choroidal thickness decreased by 49% from 241 μm to 123 μm, and fundus depigmentation, poliosis of the eyebrows and eyelashes, and skin depigmentation patches were observed.
gp100 is also expressed in normal choroidal melanocytes and is thought to be related to the mechanism of choroidal thinning 7). Ocular adverse effects may be irreversible, and regular ophthalmic monitoring is necessary during administration.
Currently, evidence for adjuvant systemic therapy is insufficient, and administration outside the framework of clinical trials is not recommended14). Families with BAP1 germline mutations (BAP1 tumor predisposition syndrome) are associated with an increased risk of multiple cancers (e.g., renal cell carcinoma, mesothelioma, cutaneous melanoma) and are candidates for genetic counseling14).
Wagle et al. (2022) reported a case of uveal melanoma tumor necrosis following COVID-19 vaccination 12). Necrotic uveal melanoma accounts for 3–6% of all cases and may present diagnostic challenges on pathology.
Combination therapy with a Gαq inhibitor (YM-254890) and a MEK inhibitor (trametinib/binimetinib) has shown synergistic antitumor effects in vitro and in vivo1). While MAPK signaling recovers within 24 hours with Gαq inhibition alone, the combination with a MEK inhibitor suppresses the recovery of MAPK signaling1).
Selumetinib: A phase II trial showed PFS improvement (vs dacarbazine/temozolomide), but the phase III trial of selumetinib plus dacarbazine (SUMIT trial) did not show PFS improvement1). MEK inhibitors have low activity rates both as monotherapy and in combination14).
Resistance mechanisms: Increased IGF1R/ROR1/2 RTK signaling, enhanced AKT signaling, and elevated GPCR (endothelin B receptor) expression have been identified, and it has been suggested that these can be overcome with pan-HDAC inhibitors1).
PKC inhibitors, decitabine (a DNA methyltransferase inhibitor), and chloroquine (an autophagy inhibitor) are also being studied as combination candidates with MEK inhibitors 1).
In uveal melanoma, the efficacy of immune checkpoint inhibitors is limited compared to cutaneous melanoma. The response rate for monotherapy is reported to be approximately 5%, and for the combination of nivolumab plus ipilimumab, it is 12–18% 14).
In the cardiac metastasis case reported by Madani et al. (2022), treatment with nivolumab 1 mg/kg plus ipilimumab 3 mg/kg (every 3 weeks for 4 cycles) followed by nivolumab maintenance therapy resulted in disease progression 6). The regimen was subsequently switched to nab-paclitaxel and temozolomide, but the outcome was ultimately poor.
Local approaches to metastatic lesions under investigation include hepatectomy, radiofrequency ablation, hepatic artery embolization, percutaneous hepatic perfusion chemotherapy (PHP) with melphalan, yttrium-90 microsphere selective internal radiation therapy (SIRT), and MR-guided laser-induced thermotherapy. PHP and SIRT show the longest overall survival 14).
The 9-ARG prognostic signature may provide insights for personalizing immunotherapy8). Detection of somatic pathogenic mutations in MBD4 is considered to potentially predict response to checkpoint inhibitors.
QWhat is Teventafusp?
A
A T-cell receptor bispecific fusion protein for HLA-A*02:01-positive patients with metastatic uveal melanoma. It targets the tumor-associated antigen gp100 and is the first drug to significantly improve overall survival in metastatic uveal melanoma in a phase III trial. During administration, attention must be paid to ocular adverse events such as choroidal thinning and fundus depigmentation.
Sriramareddy SN, Smalley KSM. MEK-ing the most of it: strategies to co-target Gαq and MAPK in uveal melanoma. Clin Cancer Res. 2021;27(5):1217-1219. doi:10.1158/1078-0432.CCR-20-4530. PMID:33355300; PMCID:PMC7925419.
Cristina Fonseca, Rita Pinto-Proença, Sabrina Bergeron, Luís Miguel Pires, Júlia Fernandes, Isabel Marques Carreira, et al. Intratumoral Heterogeneity in Uveal Melanoma. Ocul Oncol Pathol. 2020;7(1):17-25. doi:10.1159/000508517.
Ma J, Roelofs KA, Russell L, Weis E, Chen SH. Rapid growth of primary uveal melanoma following intravitreal bevacizumab injection: a case report and review of the literature. Digital journal of ophthalmology : DJO. 2021;26(3):27-30. doi:10.5693/djo.02.2020.06.001. PMID:33867879; PMCID:PMC8031910.
Jain S, Phoong KY.. Unusual presentation of a choroidal melanoma. BMJ Case Rep. 2021;14(5):e240983. doi:10.1136/bcr-2020-240983. PMID:34045197; PMCID:PMC8162134.
Tigari B, Saini M, Manchanda S, Vankdoth S. Large ciliary body melanoma. BMJ case reports. 2021;14(11). doi:10.1136/bcr-2021-246386. PMID:34764100; PMCID:PMC8587378.
Madani A, Omar NE, Mustafa G, Petkar M, Mohamed S, Al Kuwari M, Karim SA, Mohsen R.. Cardiac Metastases from Choroidal Melanoma. Clin Case Rep. 2022;10(7):e6080. doi:10.1002/ccr3.6080. PMID:35865765; PMCID:PMC9290777.
Krohn J, Vinnem LIH, Jansson RW, Straume O. Fundus hypopigmentation and choroidal thinning associated with tebentafusp therapy: report of a case and literature review. BMC ophthalmology. 2025;25(1):464. doi:10.1186/s12886-025-04274-7. PMID:40817046; PMCID:PMC12357441.
Chuah S, Chew V. Immune implication of an autophagy-related prognostic signature in uveal melanoma. Bioscience reports. 2021;41(8). doi:10.1042/BSR20211098. PMID:34374416; PMCID:PMC8380919.
Finger PT. Laser treatment for choroidal melanoma. Surv Ophthalmol. 2023;68(2):211-224.
Binkley EM, Lozano LP, Riker MJ, Pennington EC, Tucker BA, Stone EM, Boldt HC, Mullins RF.. Vascular Findings in the Choriocapillaris in a Case of Radiation Retinopathy Secondary to Choroidal Melanoma. Case Rep Ophthalmol. 2022;13(2):589-598. doi:10.1159/000525568. PMID:36160486; PMCID:PMC9459633.
Thanadar RR, Siddiqui UM, Bai S, Hou R.. Uveal Melanoma Metastasis to the Thyroid. Case Rep Endocrinol. 2023;2023:2118672. doi:10.1155/2023/2118672. PMID:37621445; PMCID:PMC10447162.