Le mélanome uvéal (uveal melanoma, UM) est une tumeur maligne provenant des mélanocytes de l’uvée (iris, corps ciliaire, choroïde). C’est la tumeur intraoculaire primitive la plus fréquente chez l’adulte, avec une origine choroïdienne dans plus de 90 % des cas, ciliaire dans environ 7 % et irienne dans 2 %. Cet article traite du mélanome de la choroïde et du corps ciliaire.
La fréquence est environ 1/20 de celle des pays occidentaux, soit 0,025 pour 100 000 habitants. Chez les Caucasiens, l’incidence est de 2 à 8 pour 1 million d’habitants, avec une légère prédominance masculine et un pic d’apparition vers 60 ans. Moins de 1 % de tous les mélanomes uvéaux surviennent avant l’âge de 18 ans. À l’échelle mondiale, environ 6 millions de personnes par an reçoivent un diagnostic de mélanome oculaire 6).
Le mode de métastase est exclusivement hématogène (en raison de l’absence de vaisseaux lymphatiques dans l’uvée), avec un fort tropisme hépatique. Des métastases hépatiques sont observées dans 70 à 90 % des cas, et les autres métastases concernent les poumons, les os et la peau 6). Plus de 62 % des métastases se manifestent cliniquement dans les 5 ans suivant le traitement de la tumeur primitive, mais les autres peuvent être détectées plus de 25 ans après. Comme les métastases peuvent apparaître plusieurs années, voire plus de 10 ans après le traitement, une surveillance à long terme est nécessaire.
La survie globale médiane après métastase, selon une méta-analyse (2494 patients, 78 articles), est de 10 à 13 mois, avec environ 2 % de survie au-delà de 5 ans et 13 % estimés survivre plus de 3 ans 13). Le taux de survie à 1 an est rapporté entre 43 et 52 %. Le taux de mortalité à 12 ans est d’environ 40 %, et ce taux est similaire quel que soit le choix du traitement local 2). Le taux de survie à 5 ans pour les tumeurs de taille moyenne est de 70 à 80 % (sans différence entre la conservation du globe et l’énucléation), et il est clair qu’il n’y a pas de différence significative dans le pronostic vital selon le traitement.
Pour les patients à risque intermédiaire à élevé, il est recommandé de poursuivre l’imagerie hépatique tous les 6 à 12 mois pendant 10 ans14).
QÀ quel point le mélanome choroïdien est-il rare ?
A
En Occident, l’incidence chez les Blancs est de 2 à 8 pour un million de personnes par an. Bien qu’il soit la tumeur intraoculaire primitive la plus fréquente chez l’adulte, le nombre absolu est faible. La fréquence est d’environ 1/20 de celle de l’Occident, soit 0,025 pour 100 000 habitants. Il est plus fréquent chez les Blancs et encore plus rare chez les Asiatiques.
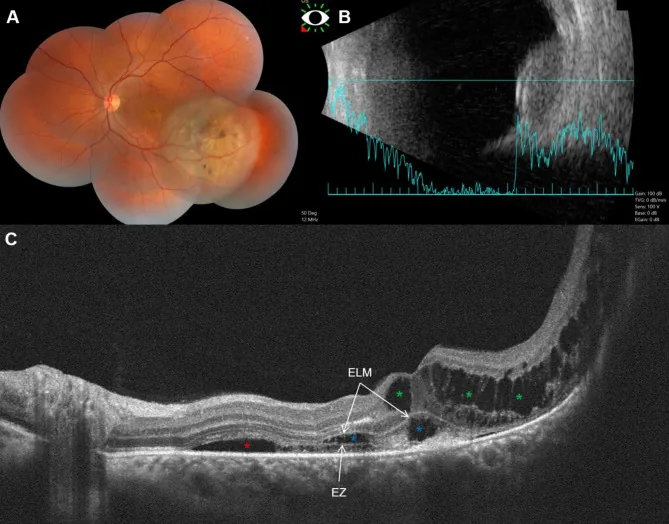
Fouad YA, et al. Bacillary layer detachment with malignant choroidal tumors: a case series. BMC Ophthalmol. 2023. Figure 1. PMCID: PMC10077734. License: CC BY.
Le panneau (A) montre une lésion surélevée non pigmentée de l’œil gauche avec pigmentation, (B) une tumeur en forme de dôme avec échogénicité interne faible à modérée, (C) un décollement sous-rétinien, un œdème kystoïde et un décollement de la membrane basale (BALAD). Cela correspond au mélanome choroïdien traité dans la section « 2. Principaux symptômes et signes cliniques ».
Environ 30 % des patients sont asymptomatiques lors de la première consultation et sont découverts fortuitement lors d’examens de routine ou d’examens pour d’autres maladies. Lorsque des symptômes apparaissent, ils se répartissent comme suit : baisse de l’acuité visuelle 38 %, photopsie 9 %, myodésopsies 7 %, déficit du champ visuel périphérique 6 %, douleur oculaire 2 %.
Lorsque la tumeur est petite et située dans la périphérie du fond d’œil, elle est généralement asymptomatique. Les symptômes suivants apparaissent avec la croissance.
Photopsie et myodésopsies : observées relativement tôt.
Déficit du champ visuel : s’étend de la périphérie vers le centre à mesure que la tumeur s’agrandit.
Métamorphopsie et baisse de l’acuité visuelle : deviennent prononcées lorsque la macula est impliquée. Un décollement séreux de la rétine survient souvent, entraînant des troubles du champ visuel et de la vision.
Hémorragie intravitréenne : peut provoquer une baisse soudaine de la vision due à un saignement de la tumeur.
Douleur oculaire : extrêmement rare, survenant dans moins de 2 % des mélanomes choroïdiens. La cause principale est un glaucome secondaire dû à une augmentation de la pression intraoculaire ou une nécrose tumorale4).
Les symptômes du mélanome ciliaire présentent les caractéristiques suivantes en raison de la particularité de sa localisation anatomique5).
Baisse indolore de l’acuité visuelle (vision floue) : plainte la plus fréquente.
Astigmatisme par déplacement du cristallin : déplacement antérieur du cristallin et du diaphragme irien dû à la tumeur.
Déficit indolore du champ visuel lors de l’invasion de l’axe visuel.
Baisse douloureuse de l’acuité visuelle due à une élévation brutale de la pression intraoculaire : secondaire à un glaucome par fermeture de l’angle.
Une élévation secondaire de la pression intraoculaire est observée chez jusqu’à 17 % des patients au moment du diagnostic de mélanome ciliaire3).
Aspect typique : apparaît unilatéralement comme une masse choroïdienne gris-brun, bombée, en forme de dôme, aux bords irréguliers.
Pigmentation : 55 % fortement pigmentés, 30 % mixtes, 15 % non pigmentés ; la teinte est variable.
Forme en champignon : environ 20 % percent la membrane de Bruch et prennent une forme de champignon (bouton de col). Croissance rapide après la perforation de la membrane de Bruch.
Liquide sous-rétinien : fréquemment associé à un décollement séreux de la rétine.
Mélanome ciliaire
Taille au moment de la découverte : souvent relativement grande en raison de la localisation anatomique cachée.
Vaisseaux sentinelles : vaisseaux épiscléraux dilatés et tortueux directement au-dessus de la tumeur, fréquemment associés.
Modifications du segment antérieur : déplacement antérieur du cristallin et du diaphragme irien, tendance à développer un glaucome secondaire par fermeture de l’angle.
Extension extraoculaire : risque d’extension extraoculaire via les canaux émissaires.
Critères de différenciation des naevus à haut risque (TFSOM-UHHD)
Pour différencier un naevus choroïdien d’un mélanome, les 8 éléments suivants sont évalués.
Épaisseur (Thickness) : supérieure à 2 mm
Liquide (Fluid) : présence de liquide sous-rétinien
Symptômes : photopsie, myodésopsies
Pigment orange : lipofuscine de couleur orange
Marge : à moins de 3 mm du disque optique
Échographie creuse (Ultrasound Hollow) : faible réflectivité interne à l’échographie
Halo absent : absence de halo
Drusen absent : absence de drusen
En l’absence de facteur de risque, la probabilité de croissance en 5 ans n’est que de 3 %, mais elle passe à 38 % avec un facteur et à plus de 50 % avec deux facteurs ou plus.
Classification de la taille tumorale selon le COMS
QComment distinguer un « grain de beauté » (nævus) choroïdien d'un mélanome ?
A
L’évaluation se fait à l’aide des 8 facteurs de risque TFSOM-UHHD. Avec 0 facteur, la probabilité de croissance en 5 ans n’est que de 3 %, mais avec 2 facteurs ou plus, elle dépasse 50 %. Une surveillance régulière par échographie et photographie du fond d’œil est nécessaire.
La survenue est principalement sporadique. La cause est inconnue, mais des anomalies des gènes suppresseurs de tumeurs ou des oncogènes, ainsi que l’exposition au soleil, sont suspectées. Les principaux facteurs de risque sont les suivants.
Couleur de l’iris claire, peau blanche, tendance à attraper des coups de soleil : fréquent chez les personnes de race blanche et d’origine nordique.
Naevus uvéal : facteur de risque le plus courant. Environ 10 % proviennent d’un naevus connu. Environ 6 % des personnes de race blanche ont un naevus choroïdien, et le taux de transformation maligne est estimé entre 1/5000 et 1/8845.
Mélanocytose oculaire congénitale, mélanocytome, neurofibromatose : facteurs de risque rares.
Antécédents familiaux de mélanome uvéal : rares mais rapportés.
Rayons ultraviolets : le rôle est considéré comme incertain.
La correspondance entre les mutations génétiques et le risque métastatique est présentée ci-dessous.
Gène muté
Fréquence de mutation
Risque métastatique / Caractéristiques
GNAQ/GNA11
83 à 89 %
Mutations initiatrices mutuellement exclusives. Aucune association directe avec le risque métastatique
BAP1
45 %
Risque métastatique le plus élevé (pic à 3,5 ans), classe 2
SF3B1
23 %
Risque modéré. Caractérisé par des métastases tardives (pic à 7 ans).
EIF1AX
17 %
Risque de métastases le plus faible.
Les mutations GNAQ/GNA11 sont considérées comme des événements précoces dans la formation tumorale ; ces mutations seules ne sont pas significativement associées à la taille de la tumeur ou au risque de métastases. Il existe une hétérogénéité intratumorale, et le profil génétique peut différer entre les sites morphologiquement distincts (par exemple, la monosomie 3 est commune à travers les sites, tandis que la délétion 6q peut être limitée aux zones pigmentées) 2).
Les mutations BAP1 sont associées au risque de métastases le plus élevé 1), tandis que les mutations EIF1AX sont associées au risque le plus faible 1).
Histologiquement, on distingue les types à cellules fusiformes et à cellules épithélioïdes. Plus la proportion de cellules épithélioïdes est élevée, plus le pronostic vital est considéré comme défavorable. Il existe également des formes mixtes.
QQuel est le rôle des mutations génétiques dans le pronostic du mélanome uvéal ?
A
Les mutations BAP1 présentent le risque de métastases le plus élevé (pic à 3,5 ans), les mutations SF3B1 sont caractérisées par des métastases tardives (pic à 7 ans), et les mutations EIF1AX indiquent le risque le plus faible. De plus, la détection d’une délétion du chromosome 3 (monosomie 3) est associée à un taux élevé de métastases et à un mauvais pronostic. Ces informations mutationnelles sont obtenues par biopsie par aspiration à l’aiguille fine et sont utilisées pour personnaliser le plan de surveillance des métastases.
Les photographies du fond d’œil en série sont extrêmement importantes pour documenter la croissance tumorale, et l’utilisation de la photographie grand angle (Optos, etc.) est également utile. En autofluorescence du fond d’œil, on utilise les propriétés d’autofluorescence de la lipofuscine pour confirmer une fluorescence orange plus brillante que les drusen (pigment orange).
L’échographie est au cœur du diagnostic du mélanome uvéal. Le mélanome malin présente une atténuation caractéristique.
Échographie A : Réflectivité interne moyenne à faible (88 %), motif décroissant (angle kappa positif), confirmation du flux sanguin intratumoral.
Échographie B : Masse surélevée en forme de dôme (le plus fréquent) ou en champignon, zone acoustiquement vide intratumorale, excavation choroïdienne, liquide sous-rétinien.
Échographie A du mélanome ciliaire : Pic élevé et réflectivité interne faible à moyenne sont caractéristiques5).
Microscopie ultrasonore biomicroscopique : Utile pour une visualisation plus détaillée du mélanome ciliaire.
Échographie Doppler couleur : Flux sanguin pulsatile à la base de la tumeur (absent dans le naevus choroïdien).
FA : Au début et au milieu de l’angiographie, on observe des vaisseaux intratumoraux et une hyperfluorescence ponctuée et en plaques multiples, et à un stade tardif, une hyperfluorescence diffuse et une fuite de colorant. L’hypofluorescence due au blocage par le pigment et l’hyperfluorescence due aux dépôts de lipofuscine au niveau de l’EPR sont mélangées.
ICGA : Les vaisseaux intratumoraux sont visualisés plus clairement (motif de double circulation). Excellente pour la visualisation des vaisseaux intratumoraux.
La SD-OCT visualise les changements de la rétine neurosensorielle et de l’EPR. Les résultats caractéristiques de l’EDI-OCT (d’après une étude de 37 yeux) sont les suivants :
Ombre choroïdienne optique (100 %)
Compression et amincissement de la choriocapillaire (100 %)
Liquide sous-rétinien (92%)
Dépôts de lipofuscine sous-rétiniens (95%)
Cellules photorécepteurs effilochées (49%) : utile pour différencier les petits mélanomes des naevus
Disparition de la zone ellipsoïde, matière hyperréflective sous-rétinienne (SRHM), décollement de la couche des bâtonnets et cônes (BALAD)
L’IRM montre un hypersignal en T1 et un hyposignal en T2. Elle est également utilisée pour le suivi de la taille tumorale après irradiation par plaque d’iode-1257).
La scintigraphie SPECT avec traceur 123I-IMP (iodoamphétamine) montre une accumulation anormale dans l’œil atteint 24 heures après injection intraveineuse, avec une sensibilité et une spécificité élevées. La TEP-FDG peut également être utilisée pour le diagnostic.
Échographie hépatique : utilisée pour le dépistage et la surveillance. Les métastases sont généralement hypoéchogènes (67%). Sensibilité élevée de 95 à 100%, sans irradiation, adaptée à une surveillance régulière13).
IRM hépatique : meilleure capacité de détection des métastases hépatiques que le scanner. Sensibilité de 83 à 100% sans irradiation13). Recommandée comme examen d’imagerie non ionisant14).
Scanner : indispensable pour le staging des métastases pulmonaires et extra-hépatiques. Bien qu’irradiant, il est essentiel pour l’évaluation initiale du staging14).
TEP-TDM : les métastases mélaniques sont hypermétaboliques au FDG. Sensibilité 94,2%, spécificité 83,3% (scanner : 55,3% et 84,4%)6).
Les tests de biomarqueurs sanguins ne sont généralement pas acceptés en dehors des tests de la fonction hépatique13).
Pour les patients à risque intermédiaire à élevé, une surveillance des métastases tous les 6 à 12 mois pendant 10 ans est recommandée14).
Les tests moléculaires sur les tissus tumoraux ou les cellules obtenues par biopsie à l’aiguille sont importants pour la stratification du risque métastatique.
Monosomie 3 (délétion du chromosome 3) : si détectée, elle entraîne un taux élevé de métastases et un mauvais pronostic. Le taux de survie à 5 ans passe d’environ 100 % à environ 50 %14).
Gain du chromosome 8q et délétion du chromosome 1p : corrélés à une diminution de la survie14).
Gain du chromosome 6p : facteur de bon pronostic (effet protecteur possible)14).
Immunohistochimie de l’expression nucléaire de BAP1 : importante pour la stratification pronostique. La perte d’expression nucléaire de BAP1 suggère une mutation de BAP1 et est associée à un risque métastatique élevé14).
LUMPO (Liverpool Uveal Melanoma Prognosticator Online) : outil de prédiction pronostique validé en externe14).
Elle est réalisée dans les cas où le diagnostic clinique est incertain. Elle est généralement effectuée en même temps que la mise en place d’une plaque de brachythérapie. Bien qu’il existe un débat en raison du risque de dissémination tumorale, elle est de plus en plus pratiquée pour la stratification pronostique par l’obtention d’un profil génétique. Compte tenu de l’hétérogénéité intratumorale, il est souhaitable de prélever plusieurs échantillons dans des zones morphologiquement différentes2).
S-100, HMB-45, MART-1 (MelanA) et la vimentine sont positifs. Pour la confirmation, il est recommandé d’utiliser au moins deux anticorps mélanocytaires en association avec la cytokératine (marqueur épithélial). Une évaluation semi-quantitative de l’indice de prolifération par Ki-67 est également réalisée. Dans le mélanome du corps ciliaire, une forte positivité pour HMB-45 a été confirmée5). La coloration nucléaire de BAP1 joue un rôle auxiliaire important dans la classification pronostique2).
Diagnostic différentiel du mélanome choroïdien : naevus choroïdien, hypertrophie congénitale de l’épithélium pigmentaire rétinien, hémorragie choroïdienne.
Diagnostic différentiel du mélanome du corps ciliaire : mélanocytome, adénome dérivé de l’épithélium pigmentaire. Une surveillance régulière par microscopie ultrasonore est importante.
Lorsque la distinction avec un naevus choroïdien n’est pas claire, une surveillance stricte est poursuivie avec des photographies du fond d’œil et une échographie en mode B. Pour un naevus choroïdien (épaisseur < 2 mm, asymptomatique), un réexamen est effectué 3 mois après l’examen initial, puis tous les 6 mois. Pour les lésions de moins de 3 mm d’épaisseur, une photographie du fond d’œil, une angiographie à la fluorescéine (FA) et une échographie en modes A et B sont réalisées initialement, avec un réexamen 3 à 4 mois plus tard, puis des photographies du fond d’œil tous les 6 à 12 mois à vie. Les petites tumeurs présentant 3 facteurs de risque ou plus doivent être traitées rapidement sans attendre les preuves de croissance.
Curiethérapie par plaque de ruthénium-106 : Une plaque contenant du ruthénium-106 (source bêta) est suturée sur la sclère au niveau de la tumeur. En raison des rayons bêta, la distance d’irradiation tissulaire est courte, ce qui convient aux tumeurs petites à moyennes. Les établissements capables de réaliser cette procédure sont limités.
Plaque I-125 : Une plaque COMS contenant de l’iode 125 est suturée sur la sclère au niveau de la tumeur. La dose d’irradiation prescrite au sommet de la tumeur est de 90 Gy. Principalement utilisé en Amérique du Nord.
Indications : Traitement de première intention pour les tumeurs de petite à moyenne taille.
Récidive : 80 % des récidives locales surviennent dans les 3 ans. 98 % sont détectables par photographie couleur du fond d’œil.
Pronostic visuel : Selon les données COMS à 3 ans, 43 à 49 % des patients ont une acuité visuelle de 20/200 (équivalent 0,1) ou moins. La rétinopathie radique en est la principale cause 10).
Protonthérapie
Caractéristiques : Thérapie par particules qui concentre la dose sur la tumeur grâce à l’effet de pic de Bragg. Traitement conservateur de l’œil.
Indications : Option de conservation oculaire pour les tumeurs de petite à moyenne taille.
Efficacité du traitement : Des cas de régression complète de la tumeur 6 mois après la protonthérapie ont été rapportés.
Avantages : Faible dose aux tissus normaux environnants.
Thérapie par faisceau de particules lourdes et CyberKnife
Les indications de la radiothérapie externe, comme la thérapie par faisceau de particules lourdes et le CyberKnife, s’élargissent. Bien que la préservation du globe oculaire soit possible, la fonction visuelle est souvent perdue en raison de complications telles que la neuropathie optique et le glaucome néovasculaire. Les établissements capables de réaliser ces traitements sont limités, mais ils sont considérés comme une option.
Traitement au laser (thermothérapie transpupillaire)
La thermothérapie transpupillaire (TTT) est indiquée pour les petits mélanomes choroïdiens (épaisseur ≤ 3 mm). Les principaux paramètres sont présentés ci-dessous 9).
Paramètre
Valeur
Longueur d’onde
810 nm (proche infrarouge)
Puissance
400–1 000 mW
Diamètre du spot
3 000 μm
Durée d’irradiation
1 à 3 minutes par spot
Profondeur
3 à 4 mm
La « thérapie sandwich » combinant thermothérapie transpupillaire et curiethérapie (plaque) rapporte un taux de récidive à 5 ans de 3 % avec l’iode-125 et de 10 % avec le ruthénium-106 9).
Lorsque la tumeur est relativement petite et surtout localisée en avant, une résection locale emportant une partie de la sclère et la tumeur seule peut être réalisée.
Elle est indiquée pour les grosses tumeurs (classification COMS : hauteur > 10 mm, plus grand diamètre basal > 16 mm) lorsque la conservation du globe est difficile. Elle reste une option aujourd’hui. En cas de récidive orbitaire après énucléation, une discussion pluridisciplinaire est nécessaire pour envisager un traitement chirurgical ou radiothérapique. La radiothérapie est recommandée à raison de 45 à 50 Gy en 20 fractions de 2 Gy.
Les options de traitement systémique pour le mélanome uvéal métastatique sont nettement plus limitées que pour le mélanome cutané.
Tébentafusp : Protéine de fusion bispécifique du récepteur des cellules T destinée aux patients HLA-A*02:01 positifs. Un essai de phase III a montré une amélioration significative de la survie globale (prolongation médiane de 6 mois par rapport au comparateur au choix de l’investigateur), ce qui en fait le premier agent efficace pour le mélanome uvéal métastatique 14). Le protocole d’administration est une perfusion IV hebdomadaire (escalade de 20 à 30 à 68 mg) 7).
Inhibiteurs de points de contrôle immunitaires : Le taux de réponse en monothérapie est d’environ 5 %, et de 12 à 18 % avec l’association nivolumab + ipilimumab. L’efficacité est limitée par rapport au mélanome cutané 14).
Inhibiteurs de MEK : Faible activité en monothérapie ou en association 14).
Traitements locaux hépatiques : Résection hépatique (si R0 possible), chimiopérfusion hépatique percutanée (PHP), radiothérapie interne sélective (SIRT), chimioembolisation artérielle hépatique (TACE) sont des options. La PHP et la SIRT montrent la survie globale la plus longue 14).
Survie globale médiane après métastase : 10 à 13 mois dans une méta-analyse, 13 % survivent plus de 3 ans 13).
La surveillance de l’œil traité est effectuée tous les 6 mois pendant 2 à 5 ans, puis une fois par an par la suite. Les cas de régression tumorale complète peuvent être transférés à un optométriste local, et les cas d’énucléation avec résection R0 peuvent être transférés à un oculariste après la cicatrisation.
Pour la surveillance des métastases, une IRM hépatique ou une échographie tous les 6 mois est recommandée pour les patients à haut risque, et une imagerie hépatique tous les 12 mois pour les patients à faible risque 14). L’imagerie non ionisante (IRM, échographie) est recommandée pour éviter l’exposition aux rayonnements. La surveillance des métastases est renforcée chez les patients à haut risque de métastases chez lesquels une monosomie 3 a été détectée.
QExiste-t-il un traitement permettant de conserver l'œil ?
A
Pour les tumeurs de petite à moyenne taille, la curiethérapie (par exemple, la suture épisclérale au 106Ru), la protonthérapie, la thérapie par ions lourds et le CyberKnife sont des options, et la conservation de l’œil est souvent possible. Pour les grosses tumeurs, l’énucléation peut être nécessaire, mais il a été démontré que le choix du traitement n’affecte pas le taux de métastases ni la survie globale. La priorité est donnée au pronostic vital tout en évaluant la possibilité de préserver la fonction visuelle.
QQuelles sont les options de traitement en cas de métastases ?
A
Si le patient est HLA-A*02:01 positif, le tebentafusp est le traitement de première intention. C’est le premier médicament à avoir montré une amélioration significative de la survie globale dans un essai de phase III. Pour les métastases hépatiques isolées, une résection hépatique est envisagée si une résection R0 est possible. Les traitements locorégionaux du foie (PHP, SIRT, TACE, etc.) sont également des options. L’efficacité des inhibiteurs de points de contrôle immunitaires est limitée par rapport au mélanome cutané (taux de réponse en monothérapie d’environ 5 %), et il est important de réaliser une réunion de concertation pluridisciplinaire dans un centre spécialisé avant de commencer le traitement.
Le mécanisme de développement du mélanome uvéal est différent de celui du mélanome cutané et emprunte des voies moléculaires distinctes.
Les mutations au niveau de la position Q209 de GNAQ/GNA11 sont les plus fréquentes ; des mutations R183 et G48L ont également été identifiées 1). Ces mutations altèrent l’activité GTPase, conduisant à un état d’activation constitutive liée au GTP 1). Les mutations GNAQ/GNA11 entraînent une activation soutenue de multiples voies de signalisation, notamment la voie MAPK (Ras/RAF/MEK/ERK) 1). Les mutations GNAQ/GNA11 sont des événements précoces dans la tumorigenèse et ne sont pas significativement associées à la taille de la tumeur ou au risque métastatique.
Les mutations secondaires (BAP1, SF3B1, EIF1AX) sont presque complètement mutuellement exclusives et ont des implications importantes pour la stratification du risque métastatique. Les mutations BAP1 sont classées dans la classe 2 (risque métastatique élevé), tandis que les porteurs de mutations SF3B1 ont une survie globale médiane après métastase de 3,9 ans (IC 95 % 2,3-6,2) et un taux de survie globale à 12 mois de 94 %, indiquant une évolution relativement favorable. Dans les métastases, GNAQ (57 %) et GNA11 (36 %) sont également détectés de manière mutuellement exclusive.
Les mutations rares initiatrices CYSLTR2 et PLCB4 sont détectées dans la quasi-totalité des mélanomes uvéaux restants.
Comme il n’y a pas de vaisseaux lymphatiques dans l’uvée, toutes les métastases se produisent par voie hématogène 6). Au début, la lésion est petite et plate, mais lorsqu’elle devient plus épaisse, elle perce la membrane de Bruch et grossit rapidement. Un décollement séreux de la rétine peut survenir autour de la tumeur.
Des cellules tumorales circulantes sont détectées chez 10 à 88 % des patients. Le tropisme marqué pour le foie est expliqué par la théorie de la « graine et du sol ». Les micrométastases peuvent survenir tôt, pendant la phase asymptomatique de la tumeur primitive. La monosomie 3 est observée dans 70 à 100 % des métastases, et les mutations BAP1 dans 60 à 80 %. Des cas de métastases dans des sites rares comme la thyroïde ont également été rapportés 11).
Deux schémas de croissance des métastases hépatiques sont connus : le type infiltrant sinusoïdal et le type nodulaire périportal. Dans le type infiltrant, la formation de pseudo-sinusoïdes par production de collagène est le mécanisme principal, tandis que dans le type nodulaire, l’angiogenèse induite par le VEGF prédomine 13).
La concentration intratumorale de VEGF est significativement plus élevée que dans les yeux sains et est positivement corrélée au diamètre basal et à la hauteur de la tumeur 3). L’administration systémique d’anti-VEGF (bévacizumab) a montré un effet inhibiteur sur les métastases dans un modèle murin, mais des résultats contradictoires ont été rapportés avec l’injection intravitréenne, qui a accéléré la croissance tumorale 3).
Un cas de croissance rapide d’un mélanome ciliaire après injection intravitréenne de bévacizumab a été rapporté (diamètre basal de 2,51 à 18,0 mm, hauteur de 6,23 à 11,0 mm en 7 semaines) 3). Le temps de doublement médian des mélanomes choroïdiens habituels est de 154 à 511 jours, cette croissance rapide est donc exceptionnelle.
Hétérogénéité intratumorale et microenvironnement immunitaire
L’hétérogénéité intratumorale existe à la fois morphologiquement et génétiquement, affectant la précision pronostique des biopsies 2). C’est pourquoi un échantillonnage multi-sites est recommandé 2).
Il a été démontré qu’une signature de neuf gènes liés à l’autophagie (9-ARG : IKBKE, BNIP1, ITGA6, FKBP1A, DLC1, PRKCD, GABARAPL1, LMCD1, TUSC1) est utile pour prédire le pronostic du mélanome uvéal (validé sur 80 cas TCGA + 150 cas GEO)8). Dans le groupe à haut risque, les voies IL6-JAK-STAT3, l’angiogenèse et les voies des espèces réactives de l’oxygène sont enrichies, et l’infiltration de cellules immunitaires (cellules T CD8+, cellules T CD4+ mémoire activées) est augmentée, mais un phénotype immunosuppresseur est associé à un mauvais pronostic, un résultat paradoxal rapporté8). Cela serait lié au fait que l’œil est un organe immunoprivilégié.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Le tébentafusp est une protéine de fusion bispécifique du récepteur des cellules T conçue pour les patients HLA-A02:01 positifs7). Il reconnaît l’antigène tumoral gp100 sur le complexe HLA-A02:01 et active les cellules T pour exercer un effet antitumoral.
Dans un essai de phase III, une amélioration significative de la survie globale (prolongation médiane de 6 mois par rapport au comparateur au choix de l’investigateur) a été démontrée chez les patients atteints de mélanome uvéal métastatique HLA-A*02:01 positifs14).
Dans un rapport de cas de Krohn et al. (2025), un patient atteint de mélanome uvéal métastatique traité par tébentafusp (IV hebdomadaire : titration 20→30→68 mg) basé sur l’essai de phase III a montré une bonne évolution avec stabilisation des métastases hépatiques et absence de nouvelles lésions après 26 mois de traitement7). Chez ce patient, l’épaisseur choroïdienne centrale de l’œil droit a diminué de 241 μm à 123 μm (amincissement de 49 %), et une dépigmentation du fond d’œil, une poliose des sourcils et des cils, et des taches de dépigmentation cutanée ont été observées.
Le gp100 est également exprimé sur les mélanocytes choroïdiens normaux, ce qui pourrait être lié au mécanisme d’amincissement choroïdien7). Les effets secondaires oculaires peuvent être irréversibles, et une surveillance ophtalmologique régulière est nécessaire pendant le traitement.
À l’heure actuelle, les preuves en faveur d’un traitement systémique adjuvant sont insuffisantes, et son administration en dehors du cadre d’un essai clinique n’est pas recommandée14). Les familles porteuses d’une mutation germinale BAP1 (syndrome de prédisposition BAP1) présentent un risque accru de plusieurs cancers (carcinome rénal, mésothéliome, mélanome cutané, etc.) et doivent bénéficier d’un conseil génétique14).
Présentation clinique rare du type nécrotique tumoral
Wagle et al. (2022) ont rapporté un cas de nécrose tumorale d’un mélanome uvéal après vaccination contre la COVID-1912). Le mélanome uvéal nécrotique représente 3 à 6 % de tous les cas et peut poser des difficultés diagnostiques.
La combinaison d’un inhibiteur de Gαq (YM-254890) et d’un inhibiteur de MEK (trametinib/binimetinib) montre un effet antitumoral synergique in vitro et in vivo1). L’inhibition seule de Gαq entraîne une récupération de la signalisation MAPK en 24 heures, mais la combinaison avec un inhibiteur de MEK supprime cette récupération1).
Sélumétinib : Un essai de phase II a montré une amélioration de la SSP (vs dacarbazine/témozolomide), mais l’essai de phase III SUMIT (sélumétinib + dacarbazine) n’a pas confirmé d’amélioration de la SSP1). Les inhibiteurs de MEK, seuls ou en combinaison, ont un faible taux d’activité14).
Mécanismes de résistance : L’augmentation de la signalisation des RTK IGF1R et ROR1/2, l’augmentation de la signalisation AKT et l’expression accrue du GPCR (récepteur de l’endothéline B) ont été identifiées, et pourraient être surmontées par les inhibiteurs de pan-HDAC1).
Les inhibiteurs de PKC, la décitabine (inhibiteur de l’ADN méthyltransférase) et la chloroquine (inhibiteur de l’autophagie) sont également étudiés comme candidats en combinaison avec les inhibiteurs de MEK1).
Dans le mélanome uvéal, l’efficacité des inhibiteurs de points de contrôle immunitaires est limitée par rapport au mélanome cutané. Le taux de réponse en monothérapie est d’environ 5 %, et de 12 à 18 % pour la combinaison nivolumab + ipilimumab14).
Dans un cas de métastase cardiaque rapporté par Madani et al. (2022), un traitement par nivolumab 1 mg/kg + ipilimumab 3 mg/kg (toutes les 3 semaines × 4 cycles) suivi d’un traitement d’entretien par nivolumab a été administré, mais la maladie a progressé6). Le traitement a ensuite été changé pour nab-paclitaxel et témozolomide, mais l’issue a finalement été défavorable.
Les approches locales pour les lésions métastatiques comprennent la résection hépatique, l’ablation par radiofréquence, l’embolisation artérielle hépatique, la perfusion hépatique percutanée (PHP) au melphalan, la radiothérapie par microsphères d’yttrium-90 (SIRT) et la thermothérapie laser induite par IRM. La PHP et la SIRT montrent la meilleure survie globale14).
Microenvironnement immunitaire et biomarqueurs pronostiques
La signature pronostique 9-ARG pourrait guider la personnalisation de l’immunothérapie8). La détection de mutations somatiques pathogènes de MBD4 pourrait prédire la réponse aux inhibiteurs de points de contrôle.
QQu'est-ce que le tebentafusp ?
A
Il s’agit d’une protéine de fusion bispécifique du récepteur des cellules T destinée aux patients atteints de mélanome uvéal métastatique HLA-A*02:01 positif. Ciblant l’antigène tumoral associé gp100, c’est le premier médicament à avoir significativement amélioré la survie globale dans le mélanome uvéal métastatique lors d’un essai de phase III. Pendant l’administration, une attention particulière doit être portée aux effets secondaires oculaires tels que l’amincissement de la choroïde et la dépigmentation du fond d’œil.
Sriramareddy SN, Smalley KSM. MEK-ing the most of it: strategies to co-target Gαq and MAPK in uveal melanoma. Clin Cancer Res. 2021;27(5):1217-1219. doi:10.1158/1078-0432.CCR-20-4530. PMID:33355300; PMCID:PMC7925419.
Cristina Fonseca, Rita Pinto-Proença, Sabrina Bergeron, Luís Miguel Pires, Júlia Fernandes, Isabel Marques Carreira, et al. Intratumoral Heterogeneity in Uveal Melanoma. Ocul Oncol Pathol. 2020;7(1):17-25. doi:10.1159/000508517.
Ma J, Roelofs KA, Russell L, Weis E, Chen SH. Rapid growth of primary uveal melanoma following intravitreal bevacizumab injection: a case report and review of the literature. Digital journal of ophthalmology : DJO. 2021;26(3):27-30. doi:10.5693/djo.02.2020.06.001. PMID:33867879; PMCID:PMC8031910.
Jain S, Phoong KY.. Unusual presentation of a choroidal melanoma. BMJ Case Rep. 2021;14(5):e240983. doi:10.1136/bcr-2020-240983. PMID:34045197; PMCID:PMC8162134.
Tigari B, Saini M, Manchanda S, Vankdoth S. Large ciliary body melanoma. BMJ case reports. 2021;14(11). doi:10.1136/bcr-2021-246386. PMID:34764100; PMCID:PMC8587378.
Madani A, Omar NE, Mustafa G, Petkar M, Mohamed S, Al Kuwari M, Karim SA, Mohsen R.. Cardiac Metastases from Choroidal Melanoma. Clin Case Rep. 2022;10(7):e6080. doi:10.1002/ccr3.6080. PMID:35865765; PMCID:PMC9290777.
Krohn J, Vinnem LIH, Jansson RW, Straume O. Fundus hypopigmentation and choroidal thinning associated with tebentafusp therapy: report of a case and literature review. BMC ophthalmology. 2025;25(1):464. doi:10.1186/s12886-025-04274-7. PMID:40817046; PMCID:PMC12357441.
Chuah S, Chew V. Immune implication of an autophagy-related prognostic signature in uveal melanoma. Bioscience reports. 2021;41(8). doi:10.1042/BSR20211098. PMID:34374416; PMCID:PMC8380919.
Finger PT. Laser treatment for choroidal melanoma. Surv Ophthalmol. 2023;68(2):211-224.
Binkley EM, Lozano LP, Riker MJ, Pennington EC, Tucker BA, Stone EM, Boldt HC, Mullins RF.. Vascular Findings in the Choriocapillaris in a Case of Radiation Retinopathy Secondary to Choroidal Melanoma. Case Rep Ophthalmol. 2022;13(2):589-598. doi:10.1159/000525568. PMID:36160486; PMCID:PMC9459633.
Thanadar RR, Siddiqui UM, Bai S, Hou R.. Uveal Melanoma Metastasis to the Thyroid. Case Rep Endocrinol. 2023;2023:2118672. doi:10.1155/2023/2118672. PMID:37621445; PMCID:PMC10447162.