Das Uveamelanom (uveal melanoma, UM) ist ein bösartiger Tumor, der aus Melanozyten der Uvea (Iris, Ziliarkörper, Aderhaut) entsteht. Es ist der häufigste primäre intraokulare Tumor bei Erwachsenen, wobei über 90 % aus der Aderhaut, etwa 7 % aus dem Ziliarkörper und 2 % aus der Iris stammen. Dieser Artikel behandelt das Aderhaut- und Ziliarkörpermelanom.
Die Häufigkeit beträgt etwa 1/20 der in westlichen Ländern, d.h. 0,025 pro 100.000 Einwohner. Bei Weißen liegt die Inzidenz bei 2–8 pro 1 Million Einwohner, mit einer leichten Bevorzugung des männlichen Geschlechts und einem Erkrankungsgipfel um das 60. Lebensjahr. Weniger als 1 % aller Uveamelanome treten vor dem 18. Lebensjahr auf. Weltweit werden jährlich etwa 6 Millionen Menschen mit einem Augenmelanom diagnostiziert 6).
Die Metastasierung erfolgt ausschließlich hämatogen (da die Uvea keine Lymphgefäße enthält) mit einer starken Leberaffinität. Lebermetastasen treten in 70–90 % der Fälle auf, weitere Metastasen betreffen Lunge, Knochen und Haut 6). Mehr als 62 % der Metastasen werden klinisch innerhalb von 5 Jahren nach Behandlung des Primärtumors manifest, die restlichen können jedoch auch nach mehr als 25 Jahren nachgewiesen werden. Da Metastasen auch Jahre bis über 10 Jahre nach der Behandlung auftreten können, ist eine langfristige Metastasenüberwachung erforderlich.
Das mediane Gesamtüberleben (OS) nach Metastasierung beträgt laut einer Metaanalyse (2494 Patienten, 78 Artikel) 10–13 Monate, wobei etwa 2 % länger als 5 Jahre und 13 % länger als 3 Jahre überleben 13). Die 1-Jahres-Überlebensrate wird mit 43–52 % angegeben. Die 12-Jahres-Mortalität beträgt etwa 40 % und ist unabhängig von der Wahl der lokalen Behandlung nahezu gleich 2). Die 5-Jahres-Überlebensrate beträgt bei mittelgroßen Tumoren 70–80 % (kein Unterschied zwischen augenerhaltender Therapie und Enukleation), und es ist klar, dass es keine großen Unterschiede in der Lebensprognose zwischen den Behandlungsmethoden gibt.
Für Patienten mit mittlerem bis hohem Risiko wird empfohlen, die Leberbildgebung 10 Jahre lang alle 6 bis 12 Monate fortzusetzen14).
QWie selten ist ein Aderhautmelanom?
A
In westlichen Ländern beträgt die Inzidenz bei Weißen 2 bis 8 pro Million Einwohner. Obwohl es der häufigste primäre intraokulare Tumor bei Erwachsenen ist, ist die absolute Zahl gering. Die Häufigkeit beträgt etwa 1/20 der westlichen Länder, also 0,025 pro 100.000 Einwohner. Es tritt häufiger bei Weißen auf und bei Asiaten noch seltener.
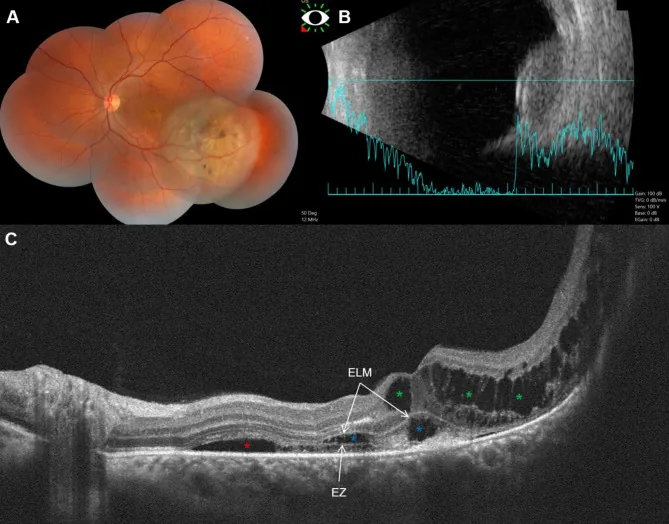
Fouad YA, et al. Bacillary layer detachment with malignant choroidal tumors: a case series. BMC Ophthalmol. 2023. Figure 1. PMCID: PMC10077734. License: CC BY.
Tafel (A) zeigt eine nicht pigmentierte erhabene Läsion des linken Auges mit Pigmentierung, (B) einen kuppelförmigen Tumor mit niedriger bis mittlerer Binnenreflexion, (C) subretinale Flüssigkeit, zystoides Ödem und Basalmembranablösung (BALAD). Dies entspricht dem Aderhautmelanom, das im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Etwa 30 % der Patienten sind beim ersten Besuch asymptomatisch und werden zufällig bei Vorsorgeuntersuchungen oder Untersuchungen anderer Erkrankungen entdeckt. Wenn Symptome auftreten, verteilen sie sich wie folgt: Sehverschlechterung 38 %, Photopsie 9 %, Mouches volantes 7 %, periphere Gesichtsfeldausfälle 6 %, Augenschmerzen 2 %.
Wenn der Tumor klein und in der Peripherie des Fundus liegt, ist er in der Regel asymptomatisch. Mit dem Wachstum treten folgende Symptome auf.
Photopsie und Mouches volantes : treten relativ früh auf.
Gesichtsfeldausfall : breitet sich mit zunehmender Tumorgröße von der Peripherie zur Mitte aus.
Metamorphopsie und Sehverschlechterung : werden deutlich, wenn die Makula betroffen ist. Häufig kommt es zu einer serösen Netzhautablösung, die Gesichtsfeld- und Sehstörungen manifestiert.
Glaskörperblutung : kann durch eine Blutung aus dem Tumor zu plötzlichem Sehverlust führen.
Augenschmerzen : extrem selten, treten bei weniger als 2 % der Aderhautmelanome auf. Hauptursachen sind ein sekundäres Glaukom aufgrund erhöhten Augeninnendrucks oder Tumornekrose4).
Die Symptome eines Ziliarkörpermelanoms zeigen aufgrund der Besonderheit seiner anatomischen Lage folgende Merkmale5).
Pigmentierung : 55 % stark pigmentiert, 30 % gemischt, 15 % unpigmentiert; die Farbe ist variabel.
Pilzförmige Gestalt : etwa 20 % durchbrechen die Bruch-Membran und nehmen eine Pilzform (Knopfform) an. Schnelles Wachstum nach Durchbruch der Bruch-Membran.
Subretinale Flüssigkeit : häufig begleitet von einer serösen Netzhautablösung.
Ziliarkörpermelanom
Größe bei Entdeckung : aufgrund der versteckten anatomischen Lage oft relativ groß bei Entdeckung.
Sentinel-Gefäße : erweiterte und geschlängelte episklerale Gefäße direkt über dem Tumor, häufig vorhanden.
Vorderabschnittsveränderungen : Vorwärtsverlagerung von Linse und Iris-Zwerchfell, Neigung zur Entwicklung eines sekundären Winkelblockglaukoms.
Extraokuläre Ausbreitung : Risiko einer extraokulären Ausbreitung über die Emissarien.
Abgrenzungskriterien für Hochrisiko-Nävi (TFSOM-UHHD)
Sonografisch hohl (Ultrasound Hollow) : niedrige Binnenreflexe im Ultraschall
Halo fehlend : Fehlen eines Halos
Drusen fehlend : Fehlen von Drusen
Bei null Risikofaktoren beträgt die Wahrscheinlichkeit eines Wachstums innerhalb von 5 Jahren nur 3 %, steigt jedoch bei einem Faktor auf 38 % und bei zwei oder mehr Faktoren auf über 50 %.
QWie unterscheidet man ein Aderhaut-„Muttermal“ (Nävus) von einem Melanom?
A
Die Bewertung erfolgt anhand der 8 Risikofaktoren TFSOM-UHHD. Bei 0 Faktoren beträgt die Wahrscheinlichkeit eines Wachstums innerhalb von 5 Jahren nur 3 %, bei 2 oder mehr Faktoren steigt sie auf über 50 %. Regelmäßige Kontrollen mittels Ultraschall und Fundusfotografie sind erforderlich.
Das Auftreten ist hauptsächlich sporadisch. Die Ursache ist unbekannt, aber es werden Anomalien von Tumorsuppressorgenen oder Onkogenen sowie Sonnenexposition vermutet. Die wichtigsten Risikofaktoren sind unten aufgeführt.
Helle Irisfarbe, helle Haut, Neigung zu Sonnenbrand : häufig bei Weißen und Menschen nordischer Abstammung.
Uveales Nävus : häufigster Risikofaktor. Etwa 10 % entstehen aus einem bekannten Nävus. Etwa 6 % der Weißen haben ein Aderhautnävus, und die Malignitätsrate wird auf 1/5000 bis 1/8845 geschätzt.
Familienanamnese von uvealem Melanom : selten, aber berichtet.
Ultraviolettes Licht : Die Rolle wird als unsicher angesehen.
Die Korrespondenz zwischen Genmutationen und Metastasierungsrisiko ist unten dargestellt.
Mutiertes Gen
Mutationshäufigkeit
Metastasierungsrisiko / Merkmale
GNAQ/GNA11
83–89 %
Gegenseitig ausschließende Initiationsmutationen. Kein direkter Zusammenhang mit Metastasierungsrisiko
BAP1
45 %
Höchstes Metastasierungsrisiko (Spitze bei 3,5 Jahren), Klasse 2
SF3B1
23 %
Mäßiges Risiko. Gekennzeichnet durch späte Metastasierung (Hauptgipfel nach 7 Jahren).
EIF1AX
17 %
Niedrigstes Metastasierungsrisiko.
GNAQ/GNA11-Mutationen gelten als frühe Ereignisse der Tumorentstehung; diese Mutationen allein sind nicht signifikant mit der Tumorgröße oder dem Metastasierungsrisiko assoziiert. Es besteht intratumorale Heterogenität, und das genetische Profil kann an morphologisch unterschiedlichen Stellen variieren (z. B. ist Monosomie 3 über alle Stellen hinweg gemeinsam, während eine 6q-Deletion auf pigmentierte Bereiche beschränkt sein kann) 2).
BAP1-Mutationen sind mit dem höchsten Metastasierungsrisiko verbunden 1), während EIF1AX-Mutationen mit dem niedrigsten verbunden sind 1).
Histologisch wird zwischen Spindelzelltyp und Epitheloidzelltyp unterschieden. Je höher der Anteil an Epitheloidzellen, desto schlechter wird die Lebensprognose eingeschätzt. Es gibt auch Mischtypen.
QWelche Rolle spielen Genmutationen bei der Prognose des Aderhautmelanoms?
A
BAP1-Mutationen weisen das höchste Metastasierungsrisiko auf (Hauptgipfel nach 3,5 Jahren), SF3B1-Mutationen sind durch späte Metastasierung (Hauptgipfel nach 7 Jahren) gekennzeichnet, und EIF1AX-Mutationen zeigen das niedrigste Risiko. Zudem ist der Nachweis einer Deletion von Chromosom 3 (Monosomie 3) mit einer hohen Metastasierungsrate und einer schlechten Prognose verbunden. Diese Mutationsinformationen werden durch Feinnadelaspirationsbiopsie gewonnen und zur Individualisierung des Metastasenüberwachungsplans verwendet.
Serielle Fundusfotografien sind für die Dokumentation des Tumorwachstums äußerst wichtig, und der Einsatz von Weitwinkel-Fundusfotografie (Optos usw.) ist ebenfalls nützlich. Bei der Fundus-Autofluoreszenz wird die Autofluoreszenz-Eigenschaft von Lipofuszin genutzt, um eine hellere orangefarbene Fluoreszenz (orangefarbenes Pigment) als Drusen zu bestätigen.
FA: In der frühen bis mittleren Phase der Angiographie zeigen sich intratumorale Gefäße und multiple punktförmige und fleckige Hyperfluoreszenzen, in der späten Phase diffuse Hyperfluoreszenz und Farbstoffleckage. Hypofluoreszenz durch Blockierung durch Pigment und Hyperfluoreszenz durch Lipofuszinablagerungen auf RPE-Ebene sind gemischt.
ICGA: Intratumorale Gefäße werden deutlicher dargestellt (Doppelkreislaufmuster). Hervorragend zur Erfassung intratumoraler Gefäße.
Die SD-OCT visualisiert Veränderungen der neurosensorischen Netzhaut und des RPE. Die charakteristischen Befunde der EDI-OCT (aus einer Studie mit 37 Augen) sind wie folgt:
Optische Aderhautverschattung (100%)
Kompression und Ausdünnung der Choriokapillaris (100%)
Subretinale Flüssigkeit (92%)
Subretinale Lipofuszinablagerungen (95%)
Ausgefranste Photorezeptoren (49%): nützlich zur Unterscheidung kleiner Melanome von Nävi
Verschwinden der Ellipsoidzone, subretinales hyperreflektives Material (SRHM), Ablösung der Stäbchen-Zapfen-Schicht (BALAD)
In T1-gewichteten Aufnahmen zeigt sich ein hohes Signal, in T2-gewichteten Aufnahmen ein niedriges Signal. Wird auch zur Nachverfolgung der Tumorgröße nach I-125-Plattenbestrahlung verwendet7).
Die SPECT-Untersuchung mit dem Tracer 123I-IMP (Iodoamphetamin) zeigt 24 Stunden nach intravenöser Injektion eine abnorme Anreicherung im betroffenen Auge und ist eine Untersuchungsmethode mit hoher Sensitivität und Spezifität. FDG-PET wird ebenfalls gelegentlich zur Diagnose eingesetzt.
Leberultraschall: Wird für Screening und Überwachung verwendet. Metastasen sind in der Regel echoarm (67%). Die Sensitivität ist mit 95–100 % hoch, und da keine Strahlenbelastung besteht, eignet er sich für regelmäßige Überwachungen13).
Leber-MRT: Bessere Erkennung von Lebermetastasen als CT. Sensitivität 83–100 %, keine Strahlenbelastung13). Wird als nichtionisierende Bildgebung empfohlen14).
CT: Unverzichtbar für das Staging von Lungen- und extrahepatischen Metastasen. Trotz Strahlenbelastung für die initiale Staging-Beurteilung erforderlich14).
Molekulare Tests an Tumorgewebe oder durch Nadelbiopsie gewonnenen Zellen sind wichtig für die Stratifizierung des Metastasierungsrisikos.
Monosomie 3 (Deletion von Chromosom 3) : Wenn nachgewiesen, führt dies zu einer hohen Metastasierungsrate und einer schlechten Prognose. Die 5-Jahres-Überlebensrate sinkt von etwa 100 % auf etwa 50 %14).
Zunahme von Chromosom 8q und Deletion von 1p : Korreliert mit verringerter Überlebensrate14).
Zunahme von Chromosom 6p : Günstiger Prognosefaktor (möglicherweise schützende Wirkung)14).
Immunhistochemie der nukleären BAP1-Expression : Wichtig für die Prognosestratifizierung. Verlust der nukleären BAP1-Expression deutet auf eine BAP1-Mutation hin und ist mit einem hohen Metastasierungsrisiko verbunden14).
Sie wird bei klinisch unsicheren Diagnosen durchgeführt. In der Regel erfolgt sie gleichzeitig mit dem Einsetzen einer Plaque-Brachytherapie. Aufgrund des Risikos einer Tumoraussaat ist sie umstritten, wird aber zunehmend zur Prognosestratifizierung durch Gewinnung eines genetischen Profils durchgeführt. Angesichts der intratumoralen Heterogenität ist eine Probenentnahme an mehreren Stellen aus morphologisch unterschiedlichen Bereichen wünschenswert2).
S-100, HMB-45, MART-1 (MelanA) und Vimentin sind positiv. Zur Bestätigung wird die Verwendung von mindestens zwei melanozytären Antikörpern in Kombination mit Zytokeratin (epithelialer Marker) empfohlen. Eine semiquantitative Bewertung des Proliferationsindex mittels Ki-67 wird ebenfalls durchgeführt. Beim Ziliarkörpermelanom wurde eine starke Positivität für HMB-45 bestätigt5). Die nukleäre BAP1-Färbung spielt eine wichtige unterstützende Rolle bei der Prognoseklassifikation2).
Differenzialdiagnose des Ziliarkörpermelanoms : Melanozytom, Adenom des Pigmentepithels. Regelmäßige Überwachung mittels Ultraschallbiomikroskopie ist wichtig.
Die Behandlungsziele sind: ① Erhalt des nutzbaren Sehvermögens des betroffenen Auges, ② Zerstörung des Tumors, ③ Verhinderung von Metastasen und Rezidiven.
Solange die Abgrenzung zum Aderhautnävus unklar ist, wird eine strenge Nachbeobachtung mit Fundusfotografie und B-Bild-Sonographie durchgeführt. Bei Aderhautnävus (Dicke < 2 mm, asymptomatisch) erfolgt eine erneute Untersuchung 3 Monate nach der Erstuntersuchung, danach alle 6 Monate. Bei Läsionen mit einer Dicke unter 3 mm werden initial Fundusfotografie, Fluoreszenzangiographie (FA) und A/B-Bild-Sonographie durchgeführt, nach 3–4 Monaten eine erneute Untersuchung, danach lebenslang alle 6–12 Monate Fundusfotografie. Kleine Tumoren mit 3 oder mehr Risikofaktoren sollten ohne Abwarten von Wachstumsnachweisen sofort behandelt werden.
106Ru-Brachytherapie mit Skleraaufnähung: Eine Plaque mit Ruthenium-106 (Betastrahler) wird auf die Sklera über dem Tumor genäht. Aufgrund der Betastrahlung ist die Gewebeeindringtiefe gering, geeignet für kleine bis mittelgroße Tumoren. Die Anzahl der Einrichtungen, die dies durchführen können, ist begrenzt.
I-125-Plaque: Eine COMS-Plaque mit Jod-125 wird auf die Sklera über dem Tumor genäht. Die verschriebene Dosis an der Tumorspitze beträgt 90 Gy. Hauptsächlich in Nordamerika verwendet.
Indikationen: Erstlinientherapie für kleine bis mittelgroße Tumoren.
Nebenwirkungen: Strahlenmakulopathie und -retinopathie, Katarakt, Neovaskularisationsglaukom, Optikusneuropathie, Skleranekrose. Die meisten treten innerhalb von 5 Jahren nach der Behandlung auf.
Rezidiv: 80 % der lokalen Rezidive treten innerhalb von 3 Jahren auf. 98 % sind mit Farbfundusfotografie nachweisbar.
Sehprognose: Nach COMS-3-Jahresdaten haben 43–49 % der Patienten einen Visus von 20/200 (entspricht 0,1) oder schlechter. Hauptursache ist die Strahlenretinopathie10).
Protonentherapie
Merkmale: Partikeltherapie, die die Dosis durch den Bragg-Peak-Effekt auf den Tumor konzentriert. Augen erhaltende Therapie.
Indikationen: Augen erhaltende Option für kleine bis mittelgroße Tumoren.
Behandlungserfolg: Es wurden Fälle einer vollständigen Tumorrückbildung 6 Monate nach Protonentherapie berichtet.
Vorteile: Geringe Dosis für das umliegende normale Gewebe.
Die Indikationen für externe Strahlentherapie wie Schwerionentherapie und CyberKnife erweitern sich. Der Augapfel kann erhalten werden, aber die Sehfunktion geht oft durch Komplikationen wie Optikusneuropathie und neovaskuläres Glaukom verloren. Die Einrichtungen, die diese Behandlungen durchführen können, sind begrenzt, aber sie werden als eine Option angesehen.
Die transpupilläre Thermotherapie (TTT) ist indiziert für kleine Aderhautmelanome (Dicke ≤ 3 mm). Die wichtigsten Parameter sind unten aufgeführt 9).
Parameter
Einstellwert
Wellenlänge
810 nm (Nahinfrarot)
Leistung
400–1.000 mW
Spotdurchmesser
3.000 μm
Bestrahlungszeit
1–3 Minuten pro Spot
Eindringtiefe
3–4 mm
Die „Sandwich-Therapie“ aus transpupillärer Thermotherapie und Brachytherapie (Plaque) zeigt eine 5-Jahres-Rezidivrate von 3 % bei Verwendung von 125I und 10 % bei 106Ru 9).
Wenn der Tumor relativ klein ist und insbesondere vorne lokalisiert ist, kann eine lokale Resektion durchgeführt werden, bei der ein Teil der Sklera und nur der Tumor entfernt werden.
Indiziert bei großen Tumoren (COMS-Großklassifikation: Höhe >10 mm, maximaler Basisdurchmesser >16 mm), wenn eine Augenerhaltung schwierig ist. Sie bleibt auch heute eine Option. Bei orbitalem Rezidiv nach Enukleation sollte in einer multidisziplinären Konferenz über chirurgische und Strahlentherapie diskutiert werden. Strahlentherapie wird mit 2 Gy/Fraktion, insgesamt 45–50 Gy in 20 Fraktionen empfohlen.
Die systemischen Behandlungsoptionen für metastasiertes Aderhautmelanom sind im Vergleich zum kutanen Melanom deutlich eingeschränkt.
Tebentafusp : T-Zell-Rezeptor-bispezifisches Fusionsprotein für HLA-A*02:01-positive Patienten. Eine Phase-III-Studie zeigte eine signifikante Verbesserung des Gesamtüberlebens (mediane Verlängerung um 6 Monate im Vergleich zur Kontrolle nach Wahl des Prüfarztes) und ist das erste wirksame Medikament für metastasiertes Aderhautmelanom14). Das Dosierungsschema ist wöchentlich i.v. (Steigerung von 20 über 30 auf 68 mg) 7).
Immun-Checkpoint-Inhibitoren : Ansprechrate als Monotherapie etwa 5 %, bei Kombination von Nivolumab und Ipilimumab 12–18 %. Die Wirksamkeit ist im Vergleich zum kutanen Melanom begrenzt 14).
MEK-Inhibitoren : Geringe Aktivität als Monotherapie oder in Kombination 14).
Lokale Lebertherapie : Leberresektion (wenn R0 möglich), perkutane hepatische Perfusionschemotherapie (PHP), selektive interne Radiotherapie (SIRT), transarterielle Chemoembolisation (TACE) sind Optionen. PHP und SIRT zeigen das längste Gesamtüberleben 14).
Medianes Gesamtüberleben nach Metastasierung: 10–13 Monate in einer Metaanalyse, 13 % überleben >3 Jahre 13).
Die Überwachung des behandelten Auges erfolgt 2–5 Jahre lang alle 6 Monate, danach einmal jährlich. Bei vollständiger Tumorrückbildung kann die Betreuung an einen örtlichen Optometristen übergeben werden, bei Enukleation mit R0-Resektion nach Wundheilung an einen Okularisten.
Zur Metastasenüberwachung wird bei Hochrisikopatienten eine Leber-MRT oder Ultraschall alle 6 Monate empfohlen, bei Niedrigrisikopatienten eine Leberbildgebung alle 12 Monate 14). Nichtionisierende Bildgebung (MRT, Ultraschall) wird zur Vermeidung von Strahlenbelastung empfohlen. Bei Patienten mit nachgewiesener Monosomie 3 und hohem Metastasenrisiko wird die Metastasenüberwachung intensiviert.
QGibt es eine Behandlung, die das Auge erhalten kann?
A
Bei kleinen bis mittelgroßen Tumoren stehen Brachytherapie (z. B. 106Ru-Sklera-Naht-Therapie), Protonentherapie, Schwerionentherapie und CyberKnife zur Verfügung, und der Augenerhalt ist oft möglich. Bei großen Tumoren kann eine Enukleation erforderlich sein, jedoch wurde gezeigt, dass die Wahl der Behandlung die Metastasierungsrate oder das Gesamtüberleben nicht beeinflusst. Die Lebensprognose hat Priorität, während die Möglichkeit des Seherhalts geprüft wird.
QWelche Behandlungsoptionen gibt es bei Metastasen?
A
Bei HLA-A*02:01-Positivität ist Tebentafusp die Erstlinientherapie. Es ist das erste Medikament, das in einer Phase-III-Studie eine signifikante Verbesserung des Gesamtüberlebens gezeigt hat. Bei leberbegrenzten Metastasen wird eine Leberresektion erwogen, wenn eine R0-Resektion möglich ist. Auch lokale Lebertherapien (PHP, SIRT, TACE usw.) sind Optionen. Die Wirksamkeit von Immun-Checkpoint-Inhibitoren ist im Vergleich zum kutanen Melanom begrenzt (Ansprechrate bei Monotherapie etwa 5 %), und vor Behandlungsbeginn sollte eine multidisziplinäre Konferenz in einem spezialisierten Zentrum stattfinden.
Der Pathogenesemechanismus des uvealen Melanoms unterscheidet sich von dem des kutanen Melanoms und verläuft über eigene molekulare Signalwege.
Mutationen an Position Q209 von GNAQ/GNA11 sind am häufigsten; auch R183- und G48L-Mutationen wurden identifiziert 1). Diese Mutationen beeinträchtigen die GTPase-Aktivität und führen zu einem konstitutiven GTP-gebundenen aktivierten Zustand 1). GNAQ/GNA11-Mutationen bewirken eine anhaltende Aktivierung mehrerer Signalwege, einschließlich des MAPK-Signalwegs (Ras/RAF/MEK/ERK) 1). GNAQ/GNA11-Mutationen sind frühe Ereignisse in der Tumorentstehung und stehen nicht in signifikantem Zusammenhang mit der Tumorgröße oder dem Metastasierungsrisiko.
Sekundäre Treibermutationen (BAP1, SF3B1, EIF1AX) treten nahezu vollständig gegenseitig exklusiv auf und haben wichtige Implikationen für die Risikostratifizierung von Metastasen. BAP1-Mutationen werden als Klasse 2 (hohes Metastasierungsrisiko) eingestuft, während Träger von SF3B1-Mutationen ein medianes Gesamtüberleben nach Metastasierung von 3,9 Jahren (95 % KI 2,3–6,2) und eine 12-Monats-Gesamtüberlebensrate von 94 % aufweisen, was auf einen relativ günstigen Verlauf hindeutet. Auch in Metastasen werden GNAQ (57 %) und GNA11 (36 %) gegenseitig exklusiv nachgewiesen.
Seltene Initiationsmutationen wie CYSLTR2 und PLCB4 werden in fast allen verbleibenden Aderhautmelanomen nachgewiesen.
Da es in der Uvea keine Lymphgefäße gibt, erfolgen alle Metastasen auf hämatogenem Weg 6). Anfangs ist die Läsion klein und flach, aber wenn sie höher wird, durchbricht sie die Bruch-Membran und wächst schnell. Es kann zu einer serösen Netzhautablösung in der Umgebung kommen.
Zirkulierende Tumorzellen werden bei 10–88 % der Patienten nachgewiesen. Die bemerkenswerte Affinität zur Leber wird durch die Seed-and-Soil-Theorie erklärt. Mikrometastasen können früh in der asymptomatischen Phase des Primärtumors auftreten. Monosomie 3 wird in 70–100 % der Metastasen gefunden, BAP1-Mutationen in 60–80 %. Auch Metastasen an seltenen Stellen wie der Schilddrüse wurden berichtet 11).
Zwei Wachstumsmuster von Lebermetastasen sind bekannt: der sinusoidale infiltrative Typ und der periportale noduläre Typ. Beim infiltrativen Typ ist die Bildung von Pseudosinusoiden durch Kollagenproduktion der Hauptmechanismus, während beim nodulären Typ die VEGF-induzierte Angiogenese vorherrscht 13).
Die intratumorale VEGF-Konzentration ist signifikant höher als in gesunden Augen und korreliert positiv mit dem Basisdurchmesser und der Höhe des Tumors 3). Die systemische Gabe von Anti-VEGF (Bevacizumab) zeigte in einem Mausmodell eine metastasierungshemmende Wirkung, aber für die intravitreale Injektion wurden widersprüchliche Ergebnisse berichtet, die das Tumorwachstum beschleunigten 3).
Ein Fall eines schnell wachsenden Ziliarkörpermelanoms nach intravitrealer Bevacizumab-Injektion wurde berichtet (Basisdurchmesser 2,51→18,0 mm, Höhe 6,23→11,0 mm in 7 Wochen) 3). Die mediane Verdopplungszeit gewöhnlicher Aderhautmelanome beträgt 154–511 Tage, daher ist dieses schnelle Wachstum ungewöhnlich.
Intratumorale Heterogenität und Immunmikroumgebung
Intratumorale Heterogenität besteht sowohl morphologisch als auch genetisch und beeinflusst die prognostische Genauigkeit von Biopsien 2). Dies ist der Grund, warum eine Probenentnahme an mehreren Stellen empfohlen wird 2).
Es wurde gezeigt, dass eine Signatur von neun Autophagie-assoziierten Genen (9-ARG: IKBKE, BNIP1, ITGA6, FKBP1A, DLC1, PRKCD, GABARAPL1, LMCD1, TUSC1) für die Prognosevorhersage des Aderhautmelanoms nützlich ist (validiert an 80 TCGA-Fällen + 150 GEO-Fällen)8). In der Hochrisikogruppe sind der IL6-JAK-STAT3-Signalweg, die Angiogenese und die Signalwege reaktiver Sauerstoffspezies angereichert, und die Infiltration von Immunzellen (CD8-T-Zellen, aktivierte Gedächtnis-CD4-T-Zellen) ist erhöht, aber ein immunsuppressiver Phänotyp ist mit einer schlechten Prognose verbunden – ein paradoxer Befund, der berichtet wurde8). Dies wird mit der Tatsache in Verbindung gebracht, dass das Auge ein immunprivilegiertes Organ ist.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Tebentafusp ist ein bispezifisches T-Zell-Rezeptor-Fusionsprotein, das für HLA-A02:01-positive Patienten entwickelt wurde7). Es erkennt das Tumor-assoziierte Antigen gp100 auf dem HLA-A02:01-Komplex und aktiviert T-Zellen, um eine antitumorale Wirkung zu entfalten.
In einer Phase-III-Studie wurde bei HLA-A*02:01-positiven Patienten mit metastasiertem Aderhautmelanom eine signifikante Verbesserung des Gesamtüberlebens (mediane Verlängerung um 6 Monate im Vergleich zur Kontrolle nach Wahl des Prüfarztes) gezeigt14).
In einem Fallbericht von Krohn et al. (2025) wurde bei einem Patienten mit metastasiertem Aderhautmelanom, der auf der Grundlage der Phase-III-Studie Tebentafusp (wöchentlich i.v.: Titration 20→30→68 mg) erhielt, nach 26-monatiger Behandlung ein guter Verlauf mit stabilen Lebermetastasen und keinen neuen Läsionen dokumentiert7). Bei demselben Patienten nahm die zentrale Aderhautdicke des rechten Auges von 241 μm auf 123 μm ab (Verdünnung um 49 %), und es wurden Fundusdepigmentierung, Poliosis der Augenbrauen und Wimpern sowie Hautdepigmentierungsflecken beobachtet.
gp100 wird auch auf normalen Aderhautmelanozyten exprimiert, was mit dem Mechanismus der Aderhautverdünnung zusammenhängen könnte7). Augenbezogene Nebenwirkungen können irreversibel sein, und während der Behandlung ist eine regelmäßige augenärztliche Überwachung erforderlich.
Derzeit ist die Evidenz für eine adjuvante systemische Therapie unzureichend, und eine Verabreichung außerhalb klinischer Studien wird nicht empfohlen14). Familien mit einer BAP1-Keimbahnmutation (BAP1-Prädispositionssyndrom) haben ein erhöhtes Risiko für mehrere Krebsarten (Nierenzellkarzinom, Mesotheliom, Hautmelanom usw.) und sollten eine genetische Beratung erhalten14).
Seltenes klinisches Bild des tumornekrotischen Typs
Wagle et al. (2022) berichteten über einen Fall von Tumornekrose eines Aderhautmelanoms nach COVID-19-Impfung12). Das nekrotische Aderhautmelanom macht 3–6 % aller Fälle aus und kann diagnostische Schwierigkeiten bereiten.
Die Kombination eines Gαq-Inhibitors (YM-254890) mit einem MEK-Inhibitor (Trametinib/Binimetinib) zeigt in vitro und in vivo synergistische antitumorale Wirkung1). Bei alleiniger Gαq-Hemmung erholt sich der MAPK-Signalweg innerhalb von 24 Stunden, während die Kombination mit einem MEK-Inhibitor die Erholung des MAPK-Signalwegs unterdrückt1).
Selumetinib: Eine Phase-II-Studie zeigte eine Verbesserung des PFS (vs. Dacarbazin/Temozolomid), aber die Phase-III-Studie SUMIT (Selumetinib + Dacarbazin) zeigte keine PFS-Verbesserung1). MEK-Inhibitoren haben sowohl als Monotherapie als auch in Kombination eine geringe Aktivitätsrate14).
Resistenzmechanismen: Erhöhte RTK-Signalgebung von IGF1R und ROR1/2, verstärkte AKT-Signalgebung und erhöhte GPCR-Expression (Endothelin-B-Rezeptor) wurden identifiziert und könnten durch pan-HDAC-Inhibitoren überwunden werden1).
PKC-Inhibitoren, Decitabin (DNA-Methyltransferase-Inhibitor) und Chloroquin (Autophagie-Inhibitor) werden ebenfalls als Kombinationskandidaten mit MEK-Inhibitoren untersucht1).
Beim Aderhautmelanom ist die Wirksamkeit von Immun-Checkpoint-Inhibitoren im Vergleich zum kutanen Melanom begrenzt. Die Ansprechrate als Monotherapie beträgt etwa 5 %, für die Kombination Nivolumab + Ipilimumab 12–18 %14).
Im Fall einer kardialen Metastase von Madani et al. (2022) wurde eine Therapie mit Nivolumab 1 mg/kg + Ipilimumab 3 mg/kg (alle 3 Wochen × 4 Zyklen) gefolgt von Nivolumab-Erhaltungstherapie durchgeführt, aber die Krankheit schritt fort6). Die Behandlung wurde später auf nab-Paclitaxel und Temozolomid umgestellt, aber das Ergebnis war letztlich ungünstig.
Zu den lokalen Ansätzen für metastatische Läsionen gehören Leberresektion, Radiofrequenzablation, Leberarterienembolisation, perkutane Leberperfusion (PHP) mit Melphalan, Yttrium-90-Mikrosphären-Brachytherapie (SIRT) und MR-gesteuerte laserinduzierte Thermotherapie. PHP und SIRT zeigen das längste Gesamtüberleben14).
Die 9-ARG-Prognosesignatur könnte Hinweise für die Personalisierung der Immuntherapie geben8). Der Nachweis somatischer pathogener MBD4-Mutationen könnte das Ansprechen auf Checkpoint-Inhibitoren vorhersagen.
QWas für ein Medikament ist Tebentafusp?
A
Es handelt sich um ein T-Zell-Rezeptor-bispezifisches Fusionsprotein für Patienten mit HLA-A*02:01-positivem metastasiertem Aderhautmelanom. Es zielt auf das tumorassoziierte Antigen gp100 ab und ist das erste Medikament, das in einer Phase-III-Studie das Gesamtüberleben bei metastasiertem Aderhautmelanom signifikant verbessert hat. Während der Verabreichung ist auf Augen-Nebenwirkungen wie Aderhautverdünnung und Fundusdepigmentierung zu achten.
Sriramareddy SN, Smalley KSM. MEK-ing the most of it: strategies to co-target Gαq and MAPK in uveal melanoma. Clin Cancer Res. 2021;27(5):1217-1219. doi:10.1158/1078-0432.CCR-20-4530. PMID:33355300; PMCID:PMC7925419.
Cristina Fonseca, Rita Pinto-Proença, Sabrina Bergeron, Luís Miguel Pires, Júlia Fernandes, Isabel Marques Carreira, et al. Intratumoral Heterogeneity in Uveal Melanoma. Ocul Oncol Pathol. 2020;7(1):17-25. doi:10.1159/000508517.
Ma J, Roelofs KA, Russell L, Weis E, Chen SH. Rapid growth of primary uveal melanoma following intravitreal bevacizumab injection: a case report and review of the literature. Digital journal of ophthalmology : DJO. 2021;26(3):27-30. doi:10.5693/djo.02.2020.06.001. PMID:33867879; PMCID:PMC8031910.
Jain S, Phoong KY.. Unusual presentation of a choroidal melanoma. BMJ Case Rep. 2021;14(5):e240983. doi:10.1136/bcr-2020-240983. PMID:34045197; PMCID:PMC8162134.
Tigari B, Saini M, Manchanda S, Vankdoth S. Large ciliary body melanoma. BMJ case reports. 2021;14(11). doi:10.1136/bcr-2021-246386. PMID:34764100; PMCID:PMC8587378.
Madani A, Omar NE, Mustafa G, Petkar M, Mohamed S, Al Kuwari M, Karim SA, Mohsen R.. Cardiac Metastases from Choroidal Melanoma. Clin Case Rep. 2022;10(7):e6080. doi:10.1002/ccr3.6080. PMID:35865765; PMCID:PMC9290777.
Krohn J, Vinnem LIH, Jansson RW, Straume O. Fundus hypopigmentation and choroidal thinning associated with tebentafusp therapy: report of a case and literature review. BMC ophthalmology. 2025;25(1):464. doi:10.1186/s12886-025-04274-7. PMID:40817046; PMCID:PMC12357441.
Chuah S, Chew V. Immune implication of an autophagy-related prognostic signature in uveal melanoma. Bioscience reports. 2021;41(8). doi:10.1042/BSR20211098. PMID:34374416; PMCID:PMC8380919.
Finger PT. Laser treatment for choroidal melanoma. Surv Ophthalmol. 2023;68(2):211-224.
Binkley EM, Lozano LP, Riker MJ, Pennington EC, Tucker BA, Stone EM, Boldt HC, Mullins RF.. Vascular Findings in the Choriocapillaris in a Case of Radiation Retinopathy Secondary to Choroidal Melanoma. Case Rep Ophthalmol. 2022;13(2):589-598. doi:10.1159/000525568. PMID:36160486; PMCID:PMC9459633.
Thanadar RR, Siddiqui UM, Bai S, Hou R.. Uveal Melanoma Metastasis to the Thyroid. Case Rep Endocrinol. 2023;2023:2118672. doi:10.1155/2023/2118672. PMID:37621445; PMCID:PMC10447162.