先天性併發症(出生時即有)

無虹彩症(Aniridia)
1. 什麼是無虹膜症
Section titled “1. 什麼是無虹膜症”無虹膜症(aniridia)是因先天性因素導致虹膜完全或不完全缺損的狀態。雖稱為「無虹膜」,但前房角最周邊處常殘留虹膜根部。
2017年依日本厚生勞動省難病法被認定為指定難病1)。經診斷為指定難病且嚴重度分級達Ⅲ級以上者,可申請醫療費用補助,並依所得設定自付額上限2)。
| 項目 | 內容 |
|---|---|
| 盛行率 | 每6.4萬至9.6萬人中1人1) |
| 性別差異 | 無1) |
| 雙眼性 | 60~90%1) |
| 遺傳模式(家族性) | 約佔總數的2/3(體染色體顯性遺傳) |
| 散發性 | 約佔總數的1/3 |
| 合併Wilms腫瘤(散發病例) | 約30%(WAGR症候群)3) |
瑞典和挪威的流行病學研究報告患病率約為每90,000人中有1人3)。對43例具有PAX6基因突變的患者進行詳細眼科評估顯示,虹膜發育異常的程度因突變類型而異3)。
Q
無虹膜症會遺傳嗎?
A
整體約2/3為體染色體顯性遺傳,患病父母有50%機率遺傳給子女。其餘1/3為散發性,無家族史。散發病例有合併Wilms腫瘤(腎腫瘤)的WAGR症候群風險,因此建議進行PAX6基因和WT1基因的遺傳學檢測。
2. 主要症狀與臨床表現
Section titled “2. 主要症狀與臨床表現”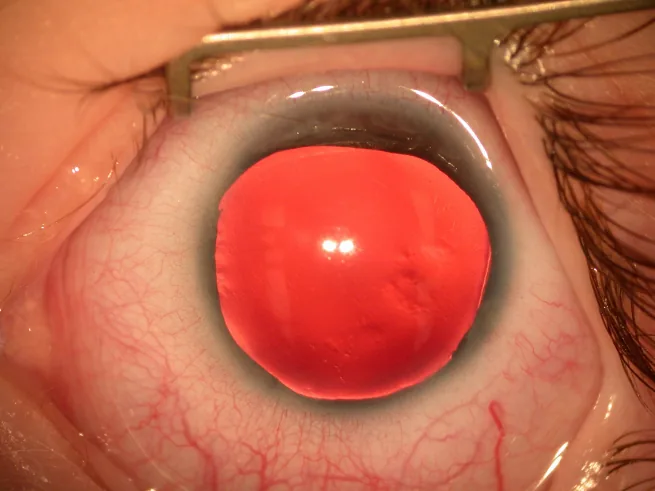
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
因虹膜缺損或不完整,瞳孔無法正常運作,無法調節進入眼球的光量,因此患者會抱怨強烈的畏光。此外,黃斑部發育不全導致的固視不良,常成為出生後早期出現的水平眼震的主訴。
後天性併發症(隨成長而發生)
眼併發症頻率總結
Section titled “眼併發症頻率總結”| 併發症 | 頻率・時期 | 對視功能的影響 |
|---|---|---|
| 黃斑部發育不全 | 幾乎全部(先天性) | 最大視力限制因素。無有效治療方法 |
| 白內障 | 約80%(後天性)1) | 視力下降・畏光惡化 |
| 青光眼 | 50〜75%(後天性)1) | 進展則造成不可逆的視野缺損 |
| 角膜緣幹細胞缺乏症 | 生長後發病並進展3) | 角膜實質混濁→嚴重視力下降 |
| 眼球震顫 | 先天性(幾乎所有病例) | 固視不良 |
| 斜視 | 先天性至嬰幼兒期 | 弱視風險 |
PAX6基因除了在眼部組織表現外,也在中樞神經、胰臟蘭氏小島及嗅上皮表現。這些組織的發育不全可能導致多種眼外併發症1)。
- 胼胝體缺損、癲癇、高階腦功能障礙
- 無嗅覺症
- 葡萄糖不耐受
- WAGR症候群(約30%的散發病例):威爾姆氏腫瘤、無虹膜、泌尿生殖器異常、智能障礙3)
Q
無虹膜症患者能看見多少?
A
視力預後通常不佳,多為0.1左右。但根據黃斑部發育不全的程度及有無併發症,個體差異可達0.1至0.7。黃斑部發育不全目前尚無有效治療方法,是視力受限的最大因素。適當的屈光矯正與低視力保健可改善生活品質。
3. 原因與風險因素
Section titled “3. 原因與風險因素”病因:PAX6基因單倍劑量不足
Section titled “病因:PAX6基因單倍劑量不足”無虹膜症的原因為位於第11號染色體短臂(11p13)的PAX6基因單一對偶基因功能喪失(單倍劑量不足)所致。功能性基因劑量減半導致此症。若兩個對偶基因皆異常,則可能導致胚胎致死1)。
PAX6是胚胎期器官分化的轉錄因子主控基因,調控多種轉錄因子。PAX6異常會引起眼球整體的多種先天性異常(如無虹膜、彼得氏異常、黃斑部發育不全等)。
基因突變類型多為無義突變、框架位移等導致早產終止密碼子(PTC)的突變,也有錯義突變的報告1)。在孤立性無虹膜症的定序分析中,約85%可檢測到PAX6突變2)。
WAGR症候群(散發病例注意事項)
Section titled “WAGR症候群(散發病例注意事項)”PAX6基因與抑癌基因WT1在11p13染色體上相鄰。散發病例可能因鄰近基因缺失而表現出威爾姆氏腫瘤、無虹膜、泌尿生殖器異常、智能障礙(mental retardation)組成的WAGR症候群3)。約30%的散發病例在5歲前會早期雙側發生威爾姆氏腫瘤。
遺傳諮詢要點
Section titled “遺傳諮詢要點”- PAX6突變陽性且無WT1缺失→可推測無WAGR症候群的可能性2)
- 遺傳學檢查應結合DNA定序與MLPA/CMA進行基因組結構異常檢測2)
- 對於疑似WAGR症候群的偶發病例,建議進行遺傳學檢測2)
Q
我應該接受無虹膜症的基因檢測嗎?
A
PAX6基因檢測對於確立Definite診斷是必要的,特別是在偶發病例中,為了評估Wilms腫瘤風險,建議進行PAX6和WT1的遺傳學檢測。檢測應結合DNA定序和MLPA/CMA,並在適當的遺傳諮詢下進行。
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”診斷標準(厚勞省指定難病 2020)
Section titled “診斷標準(厚勞省指定難病 2020)”以下顯示無虹膜症的診斷標準和嚴重度分類1)的類別分類。
| 診斷類別 | 診斷標準組合 |
|---|---|
| Definite | 符合A任一項+B1+E,並排除C |
| Probable (1) | 符合A任一項+B1+F,並排除C |
| Probable (2) | 符合A任一項+B1+B2,並排除C |
| Probable (3) | 符合A任一項+B1+B3,並排除C |
| 可能 | 符合A任一項+B1,且無法完全排除C者 |
A. 症狀
B. 檢查所見
- 裂隙燈顯微鏡檢查顯示虹膜發育異常,從部分虹膜萎縮到完全虹膜缺損不等(60~90%為雙眼性)
- 眼底檢查及OCT檢查顯示黃斑部發育不全(中心凹凹陷、黃斑色素、中心凹無血管區域不明顯)
- 裂隙燈顯微鏡檢查顯示角膜緣幹細胞缺乏、角膜混濁等角膜病變
- 裂隙燈顯微鏡檢查顯示白內障(約80%合併)
- 超音波檢查、MRI、CT顯示小眼球
- 眼球震顫
- 眼壓檢查等顯示青光眼(50~75%合併)
C. 鑑別診斷(應排除的疾病)
D. PAX6基因突變相關的眼外併發症(胼胝體缺損、癲癇等)
E. PAX6基因的致病性基因突變或11p13區域缺失(遺傳學檢查)
F. 家族內發病(約2/3為體染色體顯性遺傳)
標準檢查方法
Section titled “標準檢查方法”| 檢查 | 目的與內容 |
|---|---|
| 裂隙燈顯微鏡檢查 | 評估虹膜發育異常的程度(診斷基礎) |
| 眼底檢查與OCT | 評估黃斑部發育不全(中心凹消失、黃斑色素不明顯) |
| 隅角鏡檢查 | 評估隅角發育不全、殘留虹膜根部與小樑網的粘連 |
| 眼壓測量(定期) | 青光眼篩檢。從青少年時期定期進行 |
| 腹部超音波檢查 | Wilms腫瘤篩檢(散發病例,每隔數月,尤其5歲前) |
| 基因檢測 | 鑑定PAX6基因突變或11p13區域缺失(確診所需) |
兒童可能需要全身麻醉下檢查。
Q
無虹膜症的診斷如何進行?
5. 標準治療方法
Section titled “5. 標準治療方法”治療整體方針
Section titled “治療整體方針”虹膜發育異常、黃斑部發育不全、小眼症、眼球震顫目前無法介入,以觀察為主。治療對象為角膜病變、白內障、青光眼、畏光、低視力2)。
治療方針概要
Section titled “治療方針概要”無虹彩症需分別管理角膜、白內障、青光眼、低視力及畏光等問題2)。
| 治療領域 | 診療方針 |
|---|---|
| 角膜實質混濁 | 角膜移植對視功能改善有限,需謹慎評估適應症 |
| 角膜上皮幹細胞缺乏症 | 考慮進行眼表面重建手術 |
| 白內障 | 根據混濁及畏光程度考慮手術 |
| 高眼壓/青光眼 | 為保存視功能應積極治療 |
| 低視力照護 | 早期導入 |
| 畏光 | 使用遮光眼鏡或隱形眼鏡等措施 |
角膜病變的治療
Section titled “角膜病變的治療”角膜實質混濁:角膜移植所能改善的視功能,因無虹膜症的併發症而有限2)。長期而言,常因青光眼惡化及逐年移植片功能不良導致視力預後不佳。角膜混濁的全層角膜移植往往無法改善視力,且需注意排斥反應率高。重症病例應充分權衡利弊後再決定是否施行。
角膜上皮幹細胞缺乏症(LSCD):應考慮手術治療2)。具體而言,異體輪部移植(KLAL)或培養口腔黏膜上皮移植(COMET)可望達到一定程度的眼表面重建3)。若合併角膜實質混濁,常需同時進行角膜移植以改善視力2)。
白內障的治療
Section titled “白內障的治療”至20歲時,50~85%的患者會發生白內障,根據混濁及畏光程度規劃白內障手術2)。
- 水晶體囊及Zinn小帶脆弱,導致手術難度較高
- 需注意術後青光眼惡化、前部纖維化症候群及水泡性角膜病變的風險2)
- 植入人工水晶體(IOL)需謹慎評估適應症3)
- 不建議白內障手術同時植入人工虹膜,因可能誘發青光眼
應充分說明手術相關風險後再進行。
青光眼的治療
Section titled “青光眼的治療”青光眼直接影響視功能預後,應積極治療2)。採取以下逐步處理方式。
- 藥物治療:注意副作用,考慮對兒童的全身影響,使用眼藥水或口服藥物降低眼壓
- 流出道重建手術:隅角切開術、小樑切開術(藥物治療無效時考慮)
- 濾過手術:小樑切除術
- 青光眼植入手術:長管手術(需設施認證)
- 睫狀體凝固術:其他治療無效時的最終手段
藥物治療常出現抗藥性,導管分流手術可能是較好的選擇4)。青光眼導致的視野缺損是不可逆的,因此早期控制眼壓是維持視功能的關鍵。
Q
無虹膜症的青光眼如何治療?
A
首先進行點眼或口服藥物治療,但多數患者對藥物有抗藥性。若效果不佳,可考慮流出道重建術(隅角切開術、小樑切開術),進一步則進行小樑切除術或長管手術(青光眼植入手術)。長管手術需設施認證。睫狀體凝固術是其他治療無效時的最終手段。定期監測眼壓至關重要。
低視力照護與畏光對策
Section titled “低視力照護與畏光對策”低視力照護與畏光對策應及早導入,以維持視功能與生活品質2)。
- 屈光矯正:以眼鏡矯正屈光異常,盡可能促進視覺發育(基本)
- 遮光眼鏡:有效減輕畏光。嚴重畏光時處方使用
- 人工虹膜隱形眼鏡:可同時改善畏光與外觀
- 使用放大鏡、弱視眼鏡、擴視機等視覺輔具
6. 病理生理學與詳細發病機制
Section titled “6. 病理生理學與詳細發病機制”PAX6基因與眼部形成
Section titled “PAX6基因與眼部形成”PAX6基因是編碼胚胎期器官分化轉錄因子的主控基因。從早期眼球開始表現,統籌各種轉錄因子。PAX6單一對偶基因功能喪失(單倍劑量不足)會導致整個眼球的先天性異常(無虹膜、彼得氏異常、黃斑部發育不全等)。
PAX6突變多為無義突變或框架位移等PTC類型,也有錯義突變的報告1)。基因型與表現型的關聯研究顯示,突變類型會影響眼科發現的嚴重程度3)。
PAX6除了眼睛之外,也在中樞神經、胰臟蘭氏小島、嗅上皮表現,各組織發育不全可能導致眼外併發症(胼胝體缺損、癲癇、嗅覺缺失、葡萄糖不耐受)1)。
青光眼的發病機制
Section titled “青光眼的發病機制”無虹膜症相關青光眼的發病機制有兩種路徑。
- 開放隅角型病態:小樑網的房水流出阻力增加
- 閉鎖隅角型病態:最周邊殘留的虹膜根部與小樑網黏連,形成一種閉鎖隅角青光眼的病態
嬰兒期出現青光眼較罕見,隨著成長在青年期逐漸發病。可能因隅角發育異常而呈開放狀態,或因閉鎖隅角而導致青光眼。
角膜輪部幹細胞缺乏症(LSCD)的病態
Section titled “角膜輪部幹細胞缺乏症(LSCD)的病態”病理學上可見角膜上皮幹細胞功能異常,導致上皮和鮑曼氏層異常,形成富含血管的角膜血管翳。從福格特柵欄發育不全進展為結膜組織侵入和角質化1)。
無虹膜症的角膜比健康者厚。幼年時角膜通常正常,但隨著成長會合併角膜實質混濁和LSCD,導致視力下降。一項14年的單中心研究(738眼)顯示,在LSCD的病因中,無虹膜症佔30.9%,為最常見原因6)。
- 視力預後通常不佳,多為0.1左右
- 黃斑部發育不全無有效治療方法,是最大的視力限制因素
- 青光眼導致的視野缺損不可逆,早期眼壓控制至關重要
- 對於散發病例,需注意Wilms腫瘤在5歲前的早期發生,並持續進行定期腹部超音波檢查
關於長期預後的研究報告指出,視力預後大致不佳,但根據併發症的種類與嚴重程度,個體間存在差異5)。
7. 最新研究與未來展望
Section titled “7. 最新研究與未來展望”遺傳學檢查的進展
Section titled “遺傳學檢查的進展”隨著次世代定序(NGS)的普及,孤立性無虹膜症中PAX6突變的檢出率約達85%2)。染色體微陣列(CMA)在檢測11p13微缺失方面比傳統染色體檢查更為敏感,有助於提高WAGR症候群的診斷準確性2)。
培養口腔黏膜上皮移植(COMET)的長期成果持續累積中2)。關於Boston type I人工角膜,短期(17至28.7個月)內有65%至93%的患者視力獲得改善,但報告指出在4.5年時,該比例下降至43.5%2)。
人工虹膜裝置與基因治療的展望
Section titled “人工虹膜裝置與基因治療的展望”HumanOptics CustomFlex ArtificialIris是一種訂製的矽膠人工虹膜裝置,有助於減輕畏光並改善外觀,但截至2024年,在日本尚未獲得核准。針對PAX6單倍劑量不足的分子標靶治療目前仍處於研究階段,尚未進入臨床應用3)。
8. 參考文獻
Section titled “8. 參考文獻”- 大家義則, 川崎諭, 西田希, 木下茂, 外園千恵, 大橋裕一, 他. 無虹彩症の診断基準および重症度分類. 日眼会誌. 2020;124:83-88.
- 厚生労働科学研究費補助金難治性疾患政策研究事業「角膜難病の標準的診断法および治療法の確立を目指した調査研究」研究班. 無虹彩症の診療ガイドライン. 日眼会誌. 2021;125:38-73.
- Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
- American Academy of Ophthalmology. Diagnosis and Management of Aniridia. EyeNet Magazine. 2014. https://www.aao.org/eyenet/article/diagnosis-management-of-aniridia
- Japanese Ophthalmological Society. Clinical practice guideline for aniridia. Jpn J Ophthalmol. 2026. doi:10.1007/s10384-025-01296-y. https://link.springer.com/article/10.1007/s10384-025-01296-y
- Hu JCW, Weissbart SB. Limbal stem cell deficiency and severe ocular surface disease: a review. Ann Eye Sci. 2023;8:35.