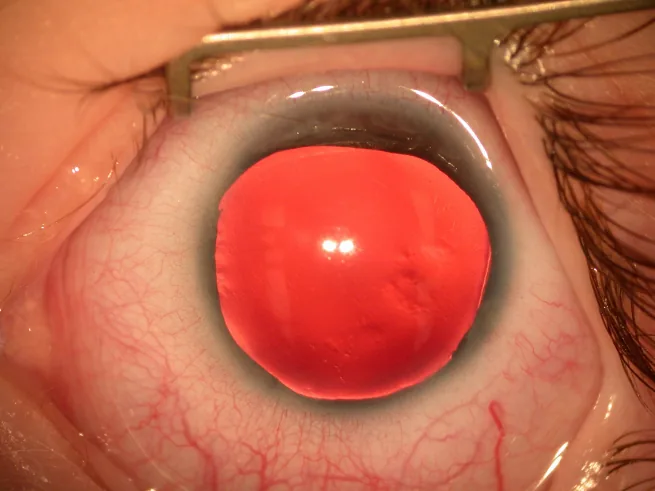
Aniridia é uma condição na qual a íris está total ou parcialmente ausente devido a fatores congênitos. Embora seja chamada de “aniridia”, frequentemente há um remanescente da raiz da íris na periferia do ângulo.
Em 2017, foi designada como doença rara com base na Lei de Doenças Raras do Ministério da Saúde, Trabalho e Bem-Estar do Japão 1). Pacientes diagnosticados com a doença designada e classificados com gravidade grau III ou superior são elegíveis para subsídio de despesas médicas, com um limite máximo de coparticipação definido de acordo com a renda 2).
Cerca de 2/3 do total (herança autossômica dominante)
Esporádico
Cerca de 1/3 do total
Associação com tumor de Wilms (casos esporádicos)
Cerca de 30% (síndrome WAGR)3)
Estudos epidemiológicos na Suécia e Noruega relatam uma prevalência de aproximadamente 1 em 90.000 pessoas3). Uma avaliação oftalmológica detalhada de 43 casos com mutações no gene PAX6 mostrou que o grau de anomalia da íris varia conforme o tipo de mutação3).
QA aniridia é hereditária?
A
Cerca de 2/3 dos casos são de herança autossômica dominante, com 50% de chance de transmissão de um pai afetado para o filho. O terço restante é esporádico, sem histórico familiar. Em casos esporádicos, há risco de síndrome WAGR, que inclui tumor de Wilms (tumor renal), sendo recomendado o teste genético dos genes PAX6 e WT1.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Fotografia com lâmpada de fenda do segmento anterior, mostrando a íris quase totalmente ausente, com apenas um fino resquício de íris na periferia. Isso ilustra diretamente o achado clínico típico da aniridia, sendo adequado para a descrição dos principais sintomas e achados clínicos.
Devido à ausência ou incompletude da íris, a pupila não funciona adequadamente, não sendo capaz de regular a quantidade de luz que entra no olho. Isso causa fotofobia intensa. Além disso, a má fixação decorrente da hipoplasia macular frequentemente leva ao nistagmo horizontal, que surge precocemente na vida.
Fotofobia: incapacidade de regular a luz pela íris → forte sensibilidade à luz
Nistagmo (nistagmo horizontal): má fixação devido à hipoplasia macular. Aparece precocemente após o nascimento
Baixa acuidade visual: fatores combinados de hipoplasia macular, catarata, glaucoma e deficiência límbica da córnea (LSCD)
Complicações congênitas (presentes desde o nascimento)
Anomalia da íris: graus variados, desde atrofia parcial até ausência completa
Hipoplasia macular: presente em quase todos os casos. Ausência da depressão foveal e pigmento macular indistinto. É o principal fator limitante da acuidade visual
Nistagmo: principalmente nistagmo horizontal. Causado por hipoplasia macular
Estrabismo: surge devido à baixa acuidade visual
Complicações adquiridas (surgem com o crescimento)
Catarata: presente em cerca de 80%. 50-85% desenvolvem até os 20 anos
Glaucoma: presente em 50-75%. Raro na infância, desenvolve-se progressivamente na adolescência
Deficiência de células-tronco do limbo (LSCD): geralmente normal na infância, mas com o crescimento, opacidade do estroma corneano e pannus vascular progridem
O gene PAX6 é expresso não apenas nos tecidos oculares, mas também no sistema nervoso central, nas ilhotas de Langerhans do pâncreas e no epitélio olfatório. A hipoplasia desses tecidos pode levar a várias complicações extraoculares1).
Agenesia do corpo caloso, epilepsia, disfunção cognitiva de ordem superior
Anosmia
Intolerância à glicose
Síndrome WAGR (cerca de 30% dos casos esporádicos): tumor de Wilms, aniridia, anomalias geniturinárias, retardo mental3)
QQuanto se enxerga com aniridia?
A
O prognóstico visual geralmente é ruim, com acuidade visual em torno de 0,1 na maioria dos casos. No entanto, dependendo do grau de hipoplasia macular e da presença de complicações, pode variar de 0,1 a 0,7. Atualmente, não há tratamento eficaz para a hipoplasia macular, sendo o principal fator limitante da visão. A correção refrativa adequada e os cuidados de baixa visão podem melhorar a qualidade de vida diária.
A aniridia é causada pela perda de função de um alelo (haploinsuficiência) do gene PAX6, localizado no braço curto do cromossomo 11 (11p13). Ocorre devido à redução pela metade da quantidade funcional do gene. Acredita-se que a anormalidade em ambos os alelos seja letal durante o desenvolvimento embrionário1).
PAX6 é um gene mestre de controle que codifica um fator de transcrição responsável pela diferenciação de órgãos durante o período embrionário, regulando vários outros fatores de transcrição. Anormalidades no PAX6 causam diversas anomalias congênitas em todo o globo ocular (aniridia, anomalia de Peters, hipoplasia macular, etc.).
Os tipos de mutações genéticas são frequentemente do tipo códon truncado prematuro (PTC), como nonsense e frameshift, e mutações missense também foram relatadas1). Na análise de sequenciamento da aniridia isolada, mutações no PAX6 são detectadas em cerca de 85% dos casos2).
O gene PAX6 está localizado próximo ao gene WT1, um gene supressor tumoral, no cromossomo 11p13. Em casos esporádicos, a deleção de genes adjacentes pode resultar na síndrome WAGR, que consiste em tumor de Wilms, aniridia, anomalias geniturinárias e retardo mental3). Cerca de 30% dos casos esporádicos desenvolvem tumor de Wilms bilateral precocemente até os 5 anos de idade.
PAX6 mutação positiva e sem deleção de WT1 → pode-se presumir que não há possibilidade de síndrome WAGR2)
O teste genético combina sequenciamento de DNA com detecção de anomalias estruturais do genoma por MLPA/CMA2)
Em casos esporádicos com suspeita de síndrome WAGR, o teste genético é recomendado 2)
QDevo fazer o teste genético para aniridia?
A
O teste genético do PAX6 é necessário para confirmar o diagnóstico definitivo, especialmente em casos esporádicos, sendo recomendado o teste genético do PAX6 e WT1 para avaliação do risco de tumor de Wilms. É importante realizar o teste combinando sequenciamento de DNA com MLPA/CMA, sob aconselhamento genético adequado.
Os critérios diagnósticos e a classificação de gravidade para aniridia1) são apresentados abaixo.
Categoria Diagnóstica
Combinação de Critérios Diagnósticos
Definitivo
Atende a qualquer um de A + B1 + E, excluindo C
Provável (1)
Atende a qualquer um de A + B1 + F, excluindo C
Provável (2)
Atende a qualquer um de A + B1 + B2, excluindo C
Provável (3)
Atende a qualquer um de A + B1 + B3, excluindo C
Possível
Atende a qualquer um dos critérios A + B1, e C não pode ser completamente excluído
A. Sintomas
Deficiência visual binocular (baixa acuidade visual devido a hipoplasia macular, catarata, glaucoma, deficiência límbica corneana)
Fotofobia (dependendo do grau de defeito da íris)
B. Achados de exames
Anomalias de formação da íris de graus variados, desde atrofia parcial até ausência completa da íris, observadas no exame com lâmpada de fenda (60-90% bilateral)
Hipoplasia macular (depressão foveal, pigmento macular e zona avascular foveal indistintos) observada em exame de fundo de olho e OCT
Triagem de glaucoma. Realizada regularmente desde a adolescência
Ultrassonografia abdominal
Triagem de tumor de Wilms (casos esporádicos, a cada poucos meses, especialmente até os 5 anos)
Teste genético
Identificação de mutação no gene PAX6 ou deleção na região 11p13 (necessário para diagnóstico definitivo)
Em crianças, pode ser necessário realizar exames sob anestesia geral.
QComo é feito o diagnóstico de aniridia?
A
O básico é confirmar a anomalia da íris com exame de lâmpada de fenda e avaliar a hipoplasia foveal com OCT. O teste genético PAX6 permite o diagnóstico definitivo, e em casos esporádicos, também se pesquisa o gene WT1. É importante diferenciar de atrofia da íris por herpes, defeito iriano pós-traumático, coloboma de íris, anomalia de Rieger e síndrome ICE.
Atualmente, não há intervenção para anomalia da íris, hipoplasia foveal, microftalmia e nistagmo, sendo o acompanhamento a base. Os alvos do tratamento são ceratopatia, catarata, glaucoma, fotofobia e baixa visão2).
Opacidade do estroma corneano: A melhora da função visual obtida pelo transplante de córnea é limitada devido às complicações da aniridia2). A longo prazo, o prognóstico visual é frequentemente ruim devido ao agravamento do glaucoma e à disfunção progressiva do enxerto. O transplante de córnea total para opacidade corneana muitas vezes não resulta em melhora visual, e deve-se atentar à alta taxa de rejeição. Em casos graves, a decisão de realizar o procedimento deve ser tomada após uma avaliação cuidadosa do equilíbrio entre benefícios e riscos.
Deficiência de células-tronco limbares (LSCD): O tratamento cirúrgico deve ser considerado 2). Especificamente, o transplante de limbo alogênico (KLAL) ou o transplante de epitélio oral cultivado (COMET) podem proporcionar alguma reconstrução da superfície ocular3). Quando há opacidade do estroma corneano associada, a combinação com transplante de córnea é frequentemente útil para melhorar a acuidade visual2).
A catarata se desenvolve em 50-85% dos pacientes até os 20 anos de idade, e a cirurgia de catarata é planejada com base na intensidade da opacidade e da fotofobia2).
Alta dificuldade cirúrgica devido à fragilidade do saco capsular e das zônulas de Zinn
Atenção aos riscos de agravamento do glaucoma pós-operatório, síndrome de fibrose anterior e ceratopatia bolhosa2)
A inserção de lente intraocular (LIO) requer indicação criteriosa 3)
A inserção de íris artificial simultânea à cirurgia de catarata não é recomendada, pois pode induzir glaucoma
O procedimento deve ser realizado após explicação adequada dos riscos cirúrgicos.
O glaucoma está diretamente relacionado ao prognóstico visual, portanto deve ser tratado ativamente 2). A abordagem é gradual, conforme abaixo.
Terapia medicamentosa: Redução da pressão intraocular com colírios e medicamentos orais, atentando para efeitos colaterais e impacto sistêmico em crianças
Cirurgia de reconstrução da via de drenagem: Goniotomia e trabeculotomia (consideradas quando a terapia medicamentosa é ineficaz)
Cirurgia de implante de glaucoma: cirurgia de tubo longo (necessita de credenciamento da instituição)
Ciclocoagulação: último recurso quando outros tratamentos não são eficazes
Há frequentemente resistência ao tratamento medicamentoso, e a cirurgia de derivação por tubo pode ser uma boa opção4). Como o glaucoma causa danos irreversíveis ao campo visual, o controle precoce da pressão intraocular é fundamental para preservar a função visual.
QComo tratar o glaucoma na aniridia?
A
Primeiro, realiza-se tratamento medicamentoso com colírios e medicamentos orais, mas muitos casos são resistentes. Se a resposta for insuficiente, considera-se a cirurgia de reconstrução da via de drenagem (goniotomia ou trabeculotomia), progredindo para trabeculectomia ou cirurgia de tubo longo (implante de glaucoma). A cirurgia de tubo longo requer credenciamento da instituição. A ciclocoagulação é o último recurso quando outros tratamentos falham. O monitoramento regular da pressão intraocular é essencial.
O gene PAX6 é um gene mestre que codifica um fator de transcrição responsável pela diferenciação de órgãos durante o período embrionário. É expresso desde o início do desenvolvimento ocular e coordena vários fatores de transcrição. A perda de função de um alelo do PAX6 (haploinsuficiência) causa anomalias congênitas em todo o olho, como aniridia, anomalia de Peters e hipoplasia macular.
Mutações no PAX6 são frequentemente do tipo PTC, como nonsense e frameshift, mas mutações missense também foram relatadas1). Estudos de correlação genótipo-fenótipo mostram que a gravidade dos achados oftalmológicos varia conforme o tipo de mutação3).
O PAX6 também é expresso fora do olho, no sistema nervoso central, nas ilhotas de Langerhans do pâncreas e no epitélio olfatório, podendo causar complicações extraoculares devido à hipoplasia desses tecidos, como agenesia do corpo caloso, epilepsia, anosmia e intolerância à glicose1).
Mecanismo de ângulo aberto: aumento da resistência ao fluxo do humor aquoso na malha trabecular
Mecanismo de ângulo fechado: aderência da raiz da íris remanescente na periferia à malha trabecular, resultando em um quadro de glaucoma de ângulo fechado
O glaucoma na infância é raro, desenvolvendo-se progressivamente na adolescência. Pode ocorrer devido a uma anomalia do ângulo que o mantém aberto ou por fechamento angular.
Patologicamente, observa-se disfunção das células-tronco do epitélio corneano, com anormalidades no epitélio e na membrana de Bowman, formando um pannus vascularizado. A hipoplasia das paliçadas de Vogt leva à invasão do tecido conjuntival e à queratinização1).
A córnea na aniridia é mais espessa que a de indivíduos saudáveis. Na infância, a córnea costuma ser normal, mas com o crescimento, ocorrem opacidade do estroma corneano e LSCD, causando perda visual. Em um estudo unicêntrico de 14 anos (738 olhos), a aniridia foi a causa mais comum de LSCD, representando 30,9% dos casos6).
O prognóstico visual geralmente é ruim, com acuidade visual em torno de 0,1
A hipoplasia macular não tem tratamento eficaz e é o principal fator limitante da visão
A perda de campo visual devido ao glaucoma é irreversível, sendo crucial o controle precoce da pressão intraocular
Em casos esporádicos, atenção ao desenvolvimento precoce do tumor de Wilms antes dos 5 anos de idade e continuar ultrassonografia abdominal regular
Estudos sobre prognóstico a longo prazo relatam que o prognóstico visual é geralmente desfavorável, mas varia individualmente conforme o tipo e a gravidade das complicações5).
Com a disseminação do sequenciamento de nova geração (NGS), a taxa de detecção de mutações no PAX6 para aniridia isolada é de aproximadamente 85%2). A microarray cromossômico (CMA) é mais sensível que os exames cromossômicos convencionais na detecção de microdeleções em 11p13, contribuindo para maior precisão diagnóstica da síndrome de WAGR2).
O acúmulo de resultados a longo prazo do transplante de mucosa oral cultivada (COMET) está avançando2). Quanto à ceratoprótese Boston tipo I, relata-se melhora visual em 65-93% a curto prazo (17-28,7 meses), mas a taxa cai para 43,5% em 4,5 anos2).
Dispositivos de íris artificial e perspectivas de terapia genética
A HumanOptics CustomFlex ArtificialIris é um dispositivo de íris artificial de silicone sob medida, considerado útil para reduzir fotofobia e melhorar a aparência, mas não é aprovado no Japão em 2024. A terapia molecular direcionada à haploinsuficiência do PAX6 está atualmente em fase de pesquisa, sem aplicação clínica3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.