L’aniridie est une condition dans laquelle l’iris est totalement ou partiellement absent en raison d’une prédisposition congénitale. Bien qu’on l’appelle « aniridie », le moignon de l’iris persiste souvent à la périphérie extrême de l’angle irido-cornéen.
Elle a été désignée comme maladie rare spécifique en vertu de la loi japonaise sur les maladies rares du ministère de la Santé, du Travail et des Affaires sociales en 20171). Les patients diagnostiqués avec cette maladie rare désignée et dont la sévérité est classée au grade III ou plus sont éligibles à l’aide médicale, avec un plafond de quote-part fixé en fonction des revenus2).
Environ 2/3 de tous les cas (autosomique dominant)
Sporadique
Environ 1/3 de tous les cas
Association à la tumeur de Wilms (cas sporadiques)
Environ 30 % (syndrome WAGR)3)
Les études épidémiologiques suédoises et norvégiennes rapportent une prévalence d’environ 1 personne sur 90 0003). Une évaluation ophtalmologique détaillée de 43 cas porteurs de mutations du gène PAX6 a montré que le degré de dysgénésie irienne varie selon le type de mutation3).
QL'aniridie est-elle héréditaire ?
A
Environ 2/3 des cas sont transmis sur le mode autosomique dominant, avec un risque de transmission de 50 % d’un parent atteint à son enfant. Le tiers restant est sporadique, sans antécédent familial. Dans les cas sporadiques, il existe un risque de syndrome WAGR associant une tumeur de Wilms (tumeur rénale), ce qui justifie un test génétique des gènes PAX6 et WT1.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
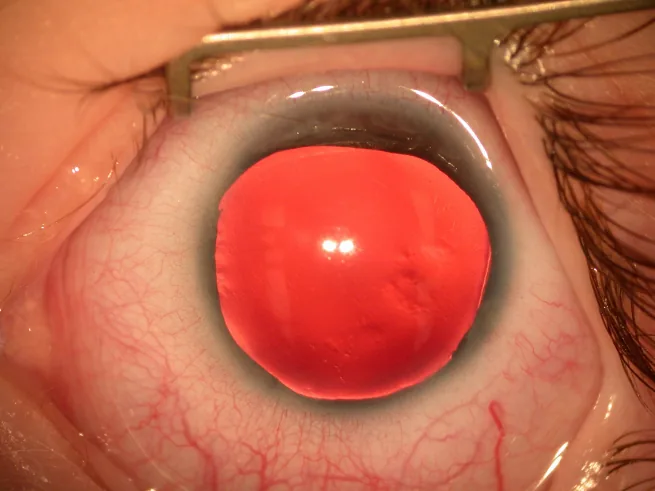
Photographie à la lampe à fente du segment antérieur montrant un iris quasi absent, avec seulement un mince vestige irien périphérique visible. Elle illustre directement les signes cliniques caractéristiques de l’aniridie et convient à la description des principaux symptômes et signes cliniques.
En raison de l’absence ou de l’incomplétude de l’iris, la pupille ne fonctionne pas et ne peut pas réguler la quantité de lumière pénétrant dans l’œil. Cela entraîne une photophobie sévère. De plus, la mauvaise fixation due à l’hypoplasie maculaire devient souvent le motif principal de consultation pour le nystagmus horizontal observé dès la petite enfance.
Photophobie : incapacité de régulation lumineuse par l’iris → éblouissement sévère
Nystagmus (nystagmus horizontal) : mauvaise fixation due à l’hypoplasie maculaire. Apparaît dès la petite enfance
Baisse de l’acuité visuelle : facteurs combinés incluant l’hypoplasie maculaire, la cataracte, le glaucome et la déficience limbique épithéliale (LSCD)
Complications congénitales (présentes dès la naissance)
Dysgénésie irienne : degrés variables allant d’une atrophie partielle à une absence complète
Hypoplasie maculaire : observée dans presque tous les cas. Disparition du creux fovéolaire et pigmentation maculaire indistincte. Principal facteur limitant l’acuité visuelle
Nystagmus : principalement horizontal. Secondaire à l’hypoplasie maculaire
Strabisme : apparaît en raison de la mauvaise vision
Complications acquises (apparaissant avec la croissance)
Cataracte : environ 80 % des cas. Survient chez 50 à 85 % des patients avant l’âge de 20 ans
Glaucome : 50 à 75 % des cas. Rare pendant la petite enfance, se développe progressivement à l’adolescence
Déficience limbique épithéliale (LSCD) : souvent normale dans la petite enfance, mais une opacification stromale cornéenne et un pannus vasculaire progressent avec la croissance
Le gène PAX6 est exprimé dans les tissus oculaires ainsi que dans le système nerveux central, les îlots de Langerhans du pancréas et l’épithélium olfactif. L’hypoplasie de ces tissus peut entraîner diverses complications extra-oculaires 1).
Agénésie du corps calleux, épilepsie, troubles des fonctions cérébrales supérieures
Anosmie
Intolérance au glucose
Syndrome WAGR (environ 30 % des cas sporadiques) : tumeur de Wilms, aniridie, anomalies urogénitales, retard mental 3)
QQuelle est l'acuité visuelle dans l'aniridie?
A
Le pronostic visuel est généralement défavorable, l’acuité visuelle étant souvent d’environ 0,1. Cependant, selon le degré d’hypoplasie maculaire et la présence ou non de complications, il existe des variations individuelles allant de 0,1 à 0,7. L’hypoplasie maculaire constitue actuellement le principal facteur limitant de l’acuité visuelle, sans traitement efficace. Une correction réfractive appropriée et des soins en basse vision peuvent améliorer la qualité de vie quotidienne.
L’aniridie est causée par une perte de fonction d’un allèle du gène PAX6 situé sur le bras court du chromosome 11 (11p13), appelée haploinsuffisance. Elle résulte d’une réduction de moitié de la dose fonctionnelle du gène. Lorsque les deux allèles sont anormaux, on pense que cela entraîne une létalité embryonnaire 1).
PAX6 est un gène maître de contrôle des facteurs de transcription qui régissent la différenciation des organes au cours de la période embryonnaire, et il coordonne divers facteurs de transcription. Les anomalies de PAX6 entraînent diverses malformations congénitales affectant l’ensemble du globe oculaire (aniridie, anomalie de Peters, hypoplasie maculaire, etc.).
Les types de mutations génétiques sont principalement des mutations de type codon tronqué prématuré (PTC) telles que les mutations non-sens et les décalages du cadre de lecture, et des mutations faux-sens ont également été rapportées 1). Dans l’analyse de séquençage de l’aniridie isolée, des mutations de PAX6 sont détectées dans environ 85 % des cas 2).
Syndrome WAGR (points d’attention pour les cas sporadiques)
Le gène PAX6 est adjacent au gène suppresseur de tumeur WT1 sur le chromosome 11p13. Dans les cas sporadiques, une délétion des gènes adjacents peut entraîner le syndrome WAGR, associant tumeur de Wilms, aniridie, anomalies urogénitales et retard mental 3). Environ 30 % des cas sporadiques développent une tumeur de Wilms bilatérale précoce avant l’âge de 5 ans.
Mutation PAX6 positive sans délétion de WT1 → le syndrome WAGR peut être considéré comme peu probable 2)
Le test génétique associe le séquençage de l’ADN à la détection d’anomalies structurelles du génome par MLPA/CMA 2)
Un test génétique est recommandé en cas de suspicion de syndrome WAGR dans les cas sporadiques2)
QFaut-il passer un test génétique pour l'aniridie ?
A
Le test génétique PAX6 est nécessaire pour confirmer le diagnostic Definite, et un test génétique PAX6 et WT1 est recommandé pour l’évaluation du risque de tumeur de Wilms, en particulier dans les cas sporadiques. Il est important de combiner le séquençage d’ADN avec MLPA/CMA et de réaliser ces tests dans le cadre d’un conseil génétique approprié.
Les critères diagnostiques de l’aniridie et la classification par catégorie selon la classification de sévérité1) sont présentés ci-dessous.
Catégorie diagnostique
Combinaison de critères diagnostiques
Definite
Répond à l’un des critères A + B1 + E, après exclusion de C
Probable (1)
Répond à l’un des critères A + B1 + F, après exclusion de C
Probable (2)
Répond à l’un des critères A + B1 + B2, après exclusion de C
Probable (3)
Répond à l’un des critères A + B1 + B3, après exclusion de C
Possible
Répond à l’un des critères A + B1, sans pouvoir exclure complètement C
A. Symptômes
Trouble visuel bilatéral (baisse de l’acuité visuelle due à une hypoplasie maculaire, une cataracte, un glaucome, une insuffisance limbique cornéenne)
Photophobie (selon le degré de défectuosité irienne)
B. Résultats d’examen
Anomalie de formation de l’iris de degré variable à l’examen à la lampe à fente, allant d’une atrophie partielle à une absence complète de l’iris (bilatérale dans 60 à 90 % des cas)
Hypoplasie maculaire au fond d’œil et à l’OCT (fovéa, pigment maculaire et zone avasculaire fovéolaire indistincts)
Lésions cornéennes telles qu’une insuffisance limbique et une opacité cornéenne à l’examen à la lampe à fente
Dépistage de la tumeur de Wilms (cas sporadiques, tous les quelques mois, surtout jusqu’à 5 ans)
Test génétique
Identification d’une mutation du gène PAX6 ou d’une délétion de la région 11p13 (nécessaire au diagnostic définitif)
Chez l’enfant, un examen sous anesthésie générale peut être nécessaire.
QComment le diagnostic d'aniridie est-il posé ?
A
L’examen à la lampe à fente permet de confirmer la dysgénésie irienne et l’OCT d’évaluer l’hypoplasie maculaire, ce qui constitue la base du diagnostic. Le test génétique PAX6 permet un diagnostic définitif, et dans les cas sporadiques, la recherche du gène WT1 est également réalisée. Le diagnostic différentiel avec l’atrophie irienne herpétique, le déficit irien post-traumatique, le colobome irien, l’anomalie de Rieger et le syndrome ICE est important.
Les anomalies de formation de l’iris, l’hypoplasie maculaire, la microphtalmie et le nystagmus ne peuvent pas être traités à l’heure actuelle ; la surveillance est la règle. Les cibles thérapeutiques sont la kératopathie, la cataracte, le glaucome, la photophobie et la basse vision2).
Opacité cornéenne stromale : l’amélioration de la fonction visuelle apportée par la greffe de cornée est limitée en raison des complications de l’aniridie2). À long terme, le pronostic visuel est souvent défavorable en raison de l’aggravation du glaucome et de la défaillance progressive du greffon. Il faut noter que la kératoplastie transfixiante pour l’opacité cornéenne n’entraîne souvent pas d’amélioration visuelle et que le taux de rejet est élevé. Dans les cas sévères, la décision d’intervention doit être prise après avoir soigneusement évalué le rapport bénéfice-risque.
Insuffisance limbique (LSCD) : envisager un traitement chirurgical2). Plus précisément, une reconstruction de la surface oculaire peut être attendue dans une certaine mesure par greffe limbique allogénique (KLAL) ou greffe de muqueuse buccale cultivée (COMET)3). En cas d’opacité stromale associée, l’ajout d’une greffe de cornée est souvent utile pour améliorer l’acuité visuelle2).
Une cataracte se développe chez 50 à 85 % des patients avant l’âge de 20 ans ; la chirurgie de la cataracte est planifiée en fonction du degré d’opacité et de photophobie2).
Difficulté chirurgicale élevée en raison de la fragilité capsulaire et des zonules de Zinn
Attention au risque d’aggravation postopératoire du glaucome, de syndrome de fibrose antérieure et de kératopathie bulleuse2)
Le glaucome ayant un impact direct sur le pronostic visuel, il doit être traité activement2). L’approche suivante est adoptée par étapes.
Traitement médicamenteux : réduire la pression intraoculaire par collyres ou voie orale, en tenant compte des effets secondaires et de l’impact systémique chez l’enfant
Chirurgie de reconstruction de la voie d’écoulement : goniotomie・trabéculotomie (envisagée si le traitement médicamenteux est inefficace)
Chirurgie d’implant de glaucome : chirurgie par tube long (nécessite un agrément d’établissement)
Cyclocoagulation : dernier recours en cas d’échec des autres traitements
La résistance au traitement médicamenteux est fréquente, et la chirurgie de shunt par tube peut être une bonne option4). Comme les atteintes du champ visuel du glaucome sont irréversibles, un contrôle précoce de la pression intraoculaire est essentiel pour préserver la fonction visuelle.
QComment traite-t-on le glaucome dans l'aniridie ?
A
Un traitement médicamenteux par collyres et voie orale est d’abord instauré, mais il est souvent résistant. En cas d’efficacité insuffisante, on envisage une chirurgie de reconstruction de la voie d’écoulement (goniotomie・trabéculotomie), puis on progresse vers une trabéculectomie ou une chirurgie par tube long (chirurgie d’implant de glaucome). La chirurgie par tube long nécessite un agrément d’établissement. La cyclocoagulation est le dernier recours en cas d’échec des autres traitements. Une surveillance régulière de la pression intraoculaire est indispensable.
Soins en basse vision et prise en charge de la photophobie
Les soins en basse vision et la prise en charge de la photophobie sont instaurés précocement pour préserver la fonction visuelle et la qualité de vie2).
Correction réfractive : corriger les troubles réfractifs avec des lunettes pour favoriser au maximum le développement visuel (de base)
Lunettes teintées : efficaces pour réduire la photophobie. Prescrites en cas de photophobie sévère
Lentilles cornéennes avec iris artificiel : utiles à la fois pour améliorer la photophobie et l’apparence esthétique
Utiliser des aides visuelles telles que les loupes, les lunettes pour amblyopes et les téléagrandisseurs
6. Physiopathologie・Mécanismes détaillés de la pathogenèse
Le gène PAX6 est un gène maître de contrôle qui code un facteur de transcription régissant la différenciation des organes embryonnaires. Il s’exprime dès le stade précoce du globe oculaire et coordonne divers facteurs de transcription. La perte de fonction d’un allèle de PAX6 (haploinsuffisance) entraîne des anomalies congénitales de l’ensemble du globe oculaire (aniridie, anomalie de Peters, hypoplasie maculaire, etc.).
Les mutations de PAX6 sont majoritairement de type PTC (non-sens・décalage du cadre de lecture), et des mutations faux-sens ont également été rapportées1). Les études sur la corrélation génotype-phénotype montrent que la sévérité des manifestations ophtalmiques varie selon le type de mutation3).
PAX6 s’exprime également en dehors de l’œil dans le système nerveux central, les îlots de Langerhans du pancréas et l’épithélium olfactif, et des complications extra-oculaires (agénésie du corps calleux・épilepsie・anosmie・intolérance au glucose) peuvent survenir en raison de l’hypoplasie de ces tissus1).
Deux voies sont envisagées dans le mécanisme de la pathogenèse du glaucome associé à l’aniridie.
Forme à angle ouvert : augmentation de la résistance à l’écoulement de l’humeur aqueuse au niveau du trabéculum
Forme à angle fermé : la racine de l’iris résiduelle dans la zone la plus périphérique adhère au trabéculum, provoquant un tableau de glaucome à angle fermé
Il est rare que le glaucome se manifeste pendant la petite enfance ; il apparaît progressivement à l’adolescence avec la croissance. Il peut survenir à angle ouvert en raison d’une dysgénésie de l’angle, ou se présenter sous forme de glaucome par fermeture de l’angle.
Physiopathologie de l’insuffisance limbique (LSCD)
Sur le plan pathologique, on observe un dysfonctionnement des cellules souches de l’épithélium cornéen, entraînant des anomalies de l’épithélium et de la membrane de Bowman, avec formation d’un pannus richement vascularisé. L’hypoplasie des palissades de Vogt évolue vers une invasion conjonctivale et une kératinisation1).
La cornée des patients atteints d’aniridie est plus épaisse que celle des sujets sains. La cornée est souvent normale pendant la petite enfance, mais avec la croissance, une opacité du stroma cornéen et une LSCD apparaissent, entraînant une baisse de l’acuité visuelle. Dans une étude monocentrique de 14 ans (738 yeux), l’aniridie représentait 30,9 % des causes de LSCD, soit la proportion la plus élevée6).
Le pronostic visuel est généralement défavorable, l’acuité visuelle étant souvent d’environ 0,1
L’hypoplasie maculaire ne bénéficie d’aucun traitement efficace et constitue le principal facteur limitant de l’acuité visuelle
Les troubles du champ visuel dus au glaucome sont irréversibles, et une gestion précoce de la pression intraoculaire est essentielle
Dans les cas sporadiques, il faut être attentif à l’apparition précoce de la tumeur de Wilms avant l’âge de 5 ans et poursuivre des échographies abdominales régulières
Les études sur le pronostic à long terme rapportent que le pronostic visuel est généralement défavorable, mais qu’il existe des variations individuelles selon le type et la sévérité des complications5).
Avec la généralisation du séquençage de nouvelle génération (NGS), le taux de détection des mutations PAX6 dans l’aniridie isolée atteint environ 85 %2). Les puces à ADN chromosomique (CMA) sont plus sensibles que les analyses chromosomiques conventionnelles pour la détection des microdélétions en 11p13 et contribuent à améliorer la précision du diagnostic du syndrome WAGR2).
Les résultats à long terme de la greffe d’épithélium buccal cultivé (COMET) s’accumulent2). Concernant la kératoprothèse de type I de Boston, une amélioration de l’acuité visuelle est obtenue chez 65 à 93 % des patients à court terme (17 à 28,7 mois), mais ce taux diminue à 43,5 % à 4,5 ans selon certaines études2).
Dispositifs d’iris artificiel et perspectives de la thérapie génique
L’HumanOptics CustomFlex ArtificialIris est un dispositif d’iris artificiel en silicone fabriqué sur mesure, considéré comme utile pour réduire la photophobie et améliorer l’apparence, mais il n’est pas approuvé au Japon à ce jour (2024). Les thérapies ciblant l’haploinsuffisance de PAX6 en sont encore au stade de la recherche et n’ont pas atteint une application clinique3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.