Les anomalies du développement du segment antérieur (ASDA), également appelées dysgénésies du segment antérieur (ASD), sont un terme générique désignant les troubles congénitaux du développement des structures du segment antérieur de l’œil, notamment la cornée, l’iris, le cristallin et l’angle de la chambre antérieure1).
Ces maladies sont extrêmement diverses sur le plan phénotypique et génétique, mais une complication importante et commune est l’augmentation de la pression intraoculaire due à une anomalie de développement de la voie d’écoulement de l’humeur aqueuse (trabéculum et canal de Schlemm), c’est-à-dire un glaucome développemental1,2).
QPourquoi les anomalies du développement du segment antérieur sont-elles souvent associées à un glaucome développemental ?
A
Le développement du segment antérieur implique des cellules mésenchymateuses dérivées de la crête neurale, qui contribuent également au développement du trabéculum et du canal de Schlemm. Lorsque des anomalies de formation du segment antérieur surviennent, la structure de la voie d’écoulement de l’humeur aqueuse est souvent également affectée, entraînant une augmentation de la résistance à l’écoulement et une élévation de la pression intraoculaire. C’est pourquoi le glaucome développemental est une complication fréquente des ASDA.
L’ARS est une maladie autosomique dominante avec des manifestations oculaires et systémiques. Les signes oculaires comprennent un embryotoxon postérieur (ligne de Schwalbe antérieure et épaissie), une hypoplasie de l’iris, une polycorie et une déviation de l’iris. On parle d’anomalie d’Axenfeld lorsque la ligne de Schwalbe proéminente adhère à l’iris, et d’anomalie de Rieger lorsqu’il y a en plus une atrophie de l’iris ou du stroma. Lorsque des anomalies osseuses ou dentaires sont associées, on parle de syndrome de Rieger. La cornée est généralement normale, de même que la structure endothéliale, mais un contact physique avec des résidus peut entraîner une opacification secondaire. Des mutations des facteurs de transcription PITX2 (chromosome 4) et FOXC1 (chromosome 6) sont en cause, et un glaucome survient dans 50 à 75 % des cas 1). Le glaucome peut apparaître dès la petite enfance, mais survient le plus souvent dans l’enfance ou chez le jeune adulte. Les phénotypes diffèrent selon la mutation : des anomalies cornéennes sont rapportées dans 50 % des cas de mutation FOXC1 et dans 16 % des cas de mutation PITX2 2).
Maladie congénitale caractérisée par une opacité cornéenne centrale et une absence de l’endothélium et de la membrane de Descemet. Elle est bilatérale dans 80 % des cas et peut s’accompagner de synéchies iridocornéennes ou de synéchies cristallinocornéennes. Des mutations de PAX6, PITX2 et CYP1B1 sont impliquées. Le syndrome de Peters-Plus associe des anomalies systémiques telles qu’une fente labiale, une petite taille et un retard mental, et est dû à des mutations du gène B3GLCT (autosomique récessif) 3). Les cas sévères nécessitent une greffe de cornée, mais le pronostic est mauvais en cas de synéchie cristallinocornéenne 3).
Maladie autosomique récessive due à une anomalie de développement du trabéculum. Les mutations du gène CYP1B1 (locus GLC3A, 2p21) sont les plus fréquentes 4). Dans une cohorte japonaise de glaucome infantile, des mutations de CYP1B1 ont été détectées dans environ 20 % des cas, et des mutations de FOXC1 ont également été rapportées 4). Au Japon, la fréquence est d’environ 1 pour 100 000 personnes ; 75 % des cas sont bilatéraux, 65 % surviennent chez les garçons et 80 % se déclarent avant l’âge d’un an. L’hypertension oculaire chez le nourrisson entraîne une augmentation du diamètre cornéen (buphtalmie), un œdème et une opacité cornéenne, et des ruptures de la membrane de Descemet (stries de Haab).
Maladie autosomique dominante caractérisée principalement par une hypoplasie de l’iris, due à des mutations du gène PAX6 (chromosome 11) 5). Outre une absence partielle ou totale de l’iris, elle peut s’accompagner d’une luxation du cristallin, d’une opacité cornéenne et d’une baisse de l’acuité visuelle due à une hypoplasie maculaire. Une délétion du gène WT1 adjacent expose au risque de syndrome WAGR (tumeur de Wilms, aniridie, anomalies génito-urinaires, retard mental), nécessitant une évaluation de WT1. En cas de délétion de WT1, une échographie rénale tous les 3 mois jusqu’à l’âge de 8 ans et un suivi en oncologie pédiatrique sont recommandés 5).
Autres maladies constitutives ①
Dystrophie endothéliale cornéenne congénitale héréditaire (CHED) : Maladie autosomique récessive caractérisée par un œdème cornéen bilatéral symétrique apparaissant à la naissance ou vers l’âge de 1 à 2 ans. Elle est causée par des mutations de SLC4A11, sans augmentation de la pression intraoculaire.
Dystrophie postérieure polymorphe de la cornée (PPCD) : Maladie autosomique dominante de l’endothélium cornéen et de la membrane de Descemet. Elle se manifeste par un œdème cornéen sans augmentation du diamètre cornéen. L’examen de l’endothélium cornéen est utile au diagnostic.
Autres maladies constitutives ②
Cornée sclérosée : Invasion non inflammatoire et non progressive du tissu scléral opaque dans la cornée périphérique, rendant la limite entre la cornée et la sclère floue. Des mutations de FOXE3, PAX6, etc., sont impliquées.
Mégalocornée : Maladie caractérisée par un diamètre cornéen ≥ 12,5 mm, souvent due à une mutation du gène CHRDL1 (hérédité récessive liée à l’X). La pression intraoculaire et les cellules endothéliales sont généralement normales. La mégalocornée congénitale se distingue du glaucome congénital primitif (PCG) par l’absence de stries de Haab et d’élargissement de l’excavation papillaire.
Les premiers symptômes du glaucome développemental sont des signes d’irritation dus à l’œdème épithélial cornéen causé par l’augmentation de la pression intraoculaire. Plus précisément, on observe un larmoiement sans sécrétion, une photophobie et un blépharospasme. Si l’hypertension oculaire persiste, l’œdème épithélial cornéen devient sévère, entraînant une opacité cornéenne. De plus, la capsule oculaire (en particulier la jonction cornée-conjonctive) s’étire, provoquant une augmentation du diamètre cornéen (buphtalmie) et une augmentation de la profondeur de la chambre antérieure.
Lorsque la cornée est étirée, la membrane de Descemet, moins élastique, se rompt (stries de Haab), et l’afflux d’humeur aqueuse dans le stroma cornéen aggrave rapidement l’œdème et l’opacité cornéens. Les stries de Haab laissent une opacité permanente et sont une cause de déficience visuelle.
Signe
Valeur normale / Référence
Diamètre cornéen du nouveau-né
9,5 à 10,5 mm (10,0 à 11,5 mm à 1 an). > 12,0 mm à la naissance suggère un PCG
Rapport cupule/disque (C/D)
Chez le nourrisson, ≥ 0,3 suggère un glaucome. Une différence interoculaire ≥ 0,2 est également évocatrice de glaucome.
Vérifier les signes du segment antérieur caractéristiques de chaque maladie. L’anneau embryonnaire postérieur (déplacement antérieur et épaississement de la ligne de Schwalbe) est associé au syndrome d’Axenfeld-Rieger (ARS) et au syndrome d’Alagille. L’hypoplasie de l’iris, la polycorie et la déviation de l’iris sont caractéristiques de l’ARS. L’opacité cornéenne centrale et les adhérences iridocornéennes suggèrent une anomalie de Peters.
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
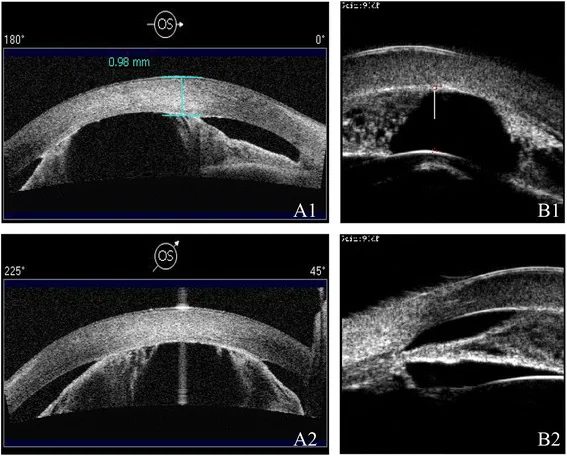
Les AS-OCT (A1/A2) et UBM (B1/B2) montrent une chambre antérieure peu profonde et des adhérences iridocornéennes avec différentes modalités. On observe un rétrécissement de la chambre antérieure et des anomalies structurelles du segment antérieur.
Selon les directives de la pratique du glaucome (5e édition) 6) et la classification internationale du Childhood Glaucoma Research Network (CGRN) 7), le glaucome pédiatrique est diagnostiqué si au moins deux des cinq critères suivants sont remplis.
Pression intraoculaire supérieure à 21 mmHg
Progression de l’augmentation du rapport cup/disc (C/D), augmentation asymétrique, ou amincissement du neuroretinal rim
Signes cornéens (stries de Haab, diamètre cornéen ≥11 mm chez le nouveau-né, ≥12 mm avant 1 an, ≥13 mm à tout âge)
Myopisation due à un allongement axial dépassant le développement normal
Déficit du champ visuel reproductible correspondant à une neuropathie optique glaucomateuse
Chez les enfants de moins de 5 ans, un examen sous sédation ou anesthésie générale est souvent nécessaire. Sous anesthésie générale, la pression intraoculaire est plus basse qu’à l’état d’éveil, il faut donc interpréter les mesures avec prudence. L’examen de l’angle se fait à l’aide d’une lampe à fente portative et d’un gonioscope direct comme la lentille de Koeppe. En cas d’opacité cornéenne rendant l’observation difficile, l’échographie biomicroscopique (UBM) est utile.
Le diagnostic du type de glaucome repose sur les signes du segment antérieur. On recherche la présence d’anneau embryotoxique postérieur, d’anomalies iriennes (ARS), de cataracte (anomalie de Peters). En cas de suspicion de cause génétique, un examen du glaucome chez les apparentés et un test génétique sont à envisager.
QQuel est le point le plus important à surveiller lors de l'examen du glaucome pédiatrique ?
A
Tous les médicaments utilisés en anesthésie générale abaissent la pression intraoculaire (la seule exception est la kétamine qui peut l’augmenter légèrement). Par conséquent, on ne peut pas exclure un glaucome sur la seule base des mesures sous anesthésie générale. Il est important de réaliser des mesures à l’état d’éveil dans la mesure du possible et d’évaluer globalement les signes autres que la pression intraoculaire, tels que le diamètre cornéen, les stries de Haab et l’excavation de la papille optique.
Le glaucome congénital précoce nécessite fondamentalement un traitement chirurgical ; le traitement médicamenteux est considéré comme un traitement à court terme pour abaisser la pression intraoculaire avant la chirurgie et comme traitement adjuvant postopératoire. Comme le patient est un nourrisson, il n’est pas rare qu’une réintervention soit nécessaire ; il est important d’expliquer clairement ce point aux parents et d’obtenir leur compréhension.
La première intervention peut être une goniotomie ou une trabéculotomie. Ces interventions agissent directement sur l’anomalie de développement de la voie d’écoulement de l’humeur aqueuse. En cas d’échec, une chirurgie filtrante ou une implantation de tube de drainage sont envisagées.
Même après le contrôle de la pression intraoculaire, un traitement de l’amblyopie est souvent nécessaire. L’anisométropie, l’astigmatisme irrégulier, l’opacité cornéenne et les stries de Haab peuvent être des causes d’amblyopie ; il est donc important de réaliser régulièrement des examens de l’acuité visuelle et de la réfraction après la chirurgie.
Dans l’anomalie de Peters avec opacité cornéenne sévère, une greffe de cornée (kératoplastie transfixiante) est indiquée, mais dans les cas graves avec adhérence cristallin-cornée, le risque d’échec du greffon est élevé.
Au cours du développement du segment antérieur, l’épithélium cornéen et le cristallin dérivés de l’ectoderme de surface, ainsi que l’épithélium pigmentaire rétinien dérivé du neuroectoderme, interagissent avec les cellules mésenchymateuses dérivées de la crête neurale pour former une structure normale du segment antérieur. Les cellules de la crête neurale participent à la formation du stroma cornéen, de l’endothélium cornéen, du trabéculum et du stroma irien. Des anomalies de migration ou de différenciation de ces cellules entraînent des dysgénésies du segment antérieur.
Dans l’ARS, FOXC1 et PITX2 interagissent physiquement et sont essentiels à la différenciation normale des cellules de la crête neurale. Les mutations de l’un ou l’autre gène peuvent produire des phénotypes similaires en raison de cette interaction.
QLes anomalies du développement du segment antérieur peuvent-elles être diagnostiquées de manière définitive par un test génétique ?
A
Pas dans tous les cas. Dans 40 à 75 % des cas, la cause génétique n’est pas encore identifiée. Cependant, les tests génétiques sont utiles pour le diagnostic différentiel (par exemple, mutation PAX6 dans l’aniridie et évaluation du gène WT1 adjacent pour exclure le syndrome WAGR), le dépistage familial et le conseil génétique. En particulier dans l’ARS, 50 à 75 % des apparentés peuvent développer un glaucome, donc un test génétique et un dépistage de la pression intraoculaire et du glaucome chez les membres de la famille sont recommandés.
QLes facteurs environnementaux sont-ils également un risque d'anomalies du développement du segment antérieur ?
A
Une étude cas-témoins coréenne (582 cas vs 1 746 témoins) a rapporté qu’une augmentation de l’exposition maternelle aux PM2,5 au cours des trois mois précédant la conception et des premier et deuxième trimestres de grossesse était associée à un risque accru d’ASD (aniridie, hypoplasie de l’iris, anomalie de Peters, ARS, PCG) chez l’enfant 8). Outre les facteurs génétiques, les facteurs environnementaux tels que les infections pendant la grossesse et les substances tératogènes peuvent également perturber le développement normal du segment antérieur.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.