Le anomalie dello sviluppo del segmento anteriore (ASDA), chiamate anche disgenesi del segmento anteriore (ASD), sono un termine generico per i disturbi congeniti dello sviluppo delle strutture del segmento anteriore dell’occhio, tra cui cornea, iride, cristallino e angolo della camera anteriore1).
Queste malattie sono estremamente eterogenee dal punto di vista fenotipico e genetico, ma una complicanza comune e importante è l’aumento della pressione intraoculare dovuto a un’anomalia dello sviluppo della via di deflusso dell’umore acqueo (trabecolato e canale di Schlemm), cioè il glaucoma evolutivo 1,2).
QPerché le anomalie dello sviluppo del segmento anteriore sono spesso associate a glaucoma evolutivo?
A
Lo sviluppo del segmento anteriore coinvolge cellule mesenchimali derivate dalla cresta neurale, che contribuiscono anche allo sviluppo del trabecolato e del canale di Schlemm. Quando si verifica un’anomalia di formazione del segmento anteriore, la struttura della via di deflusso dell’umore acqueo è spesso compromessa, portando a un aumento della resistenza al deflusso e a un innalzamento della pressione intraoculare. Pertanto, il glaucoma evolutivo è una complicanza comune delle ASDA.
L’ARS è una malattia autosomica dominante con manifestazioni oculari e sistemiche. I segni oculari includono embriotossone posteriore (linea di Schwalbe anteriorizzata e ispessita), ipoplasia dell’iride, policoria e deviazione dell’iride. Una linea di Schwalbe prominente con aderenze all’iride è chiamata anomalia di Axenfeld, mentre se si aggiunge atrofia dell’iride o dello stroma si parla di anomalia di Rieger. Quando sono presenti anomalie scheletriche o dentali, si parla di sindrome di Rieger. La cornea è generalmente normale, così come la struttura endoteliale, ma il contatto fisico con residui può causare opacità secondaria. Mutazioni dei fattori di trascrizione PITX2 (cromosoma 4) e FOXC1 (cromosoma 6) sono la causa, e nel 50-75% dei casi si sviluppa glaucoma1). Il glaucoma può insorgere nell’infanzia, ma più spesso si manifesta nell’infanzia o nella giovane età adulta. I fenotipi differiscono: con mutazioni FOXC1 si riportano anomalie corneali nel 50% dei casi, con mutazioni PITX2 nel 16% 2).
Malattia congenita caratterizzata da opacità corneale centrale e assenza di endotelio corneale e membrana di Descemet. L’80% dei casi è bilaterale e possono essere presenti sinechie iridocorneali o lenticolocorneali. Sono coinvolte mutazioni di PAX6, PITX2 e CYP1B1. La sindrome di Peters-Plus comprende anomalie sistemiche come labbro leporino, bassa statura e ritardo mentale, ed è causata da mutazioni del gene B3GLCT (autosomica recessiva) 3). I casi gravi richiedono un trapianto di cornea, ma la prognosi è sfavorevole in caso di sinechie lenticolocorneali 3).
Malattia autosomica recessiva causata da un’anomalia dello sviluppo del trabecolato. Le mutazioni del gene CYP1B1 (locus GLC3A, 2p21) sono le più frequenti 4). In una coorte giapponese di glaucoma pediatrico, mutazioni di CYP1B1 sono state rilevate in circa il 20% dei casi, e sono state riportate anche mutazioni di FOXC1 4). In Giappone la frequenza è di circa 1 su 100.000; il 75% dei casi è bilaterale, il 65% si verifica nei maschi e l’80% si manifesta entro il primo anno di vita. L’ipertensione oculare nei neonati causa un aumento del diametro corneale (buftalmo), edema e opacità corneale, e rotture della membrana di Descemet (strie di Haab).
Malattia autosomica dominante caratterizzata principalmente da ipoplasia dell’iride, causata da mutazioni del gene PAX6 (cromosoma 11) 5). Oltre all’assenza parziale o totale dell’iride, possono verificarsi lussazione del cristallino, opacità corneale e deficit visivo da ipoplasia maculare. Una delezione del gene WT1 adiacente comporta il rischio di sindrome WAGR (tumore di Wilms, aniridia, anomalie genitourinarie, ritardo mentale), rendendo necessaria la valutazione di WT1. In caso di delezione di WT1, si raccomanda ecografia renale ogni 3 mesi fino agli 8 anni e follow-up oncologico pediatrico 5).
Altre malattie costituenti ①
Distrofia endoteliale corneale congenita ereditaria (CHED) : Malattia autosomica recessiva caratterizzata da edema corneale bilaterale simmetrico che compare alla nascita o intorno a 1-2 anni di età. Causata da mutazioni di SLC4A11, senza aumento della pressione intraoculare.
Distrofia corneale posteriore polimorfa (PPCD) : Malattia autosomica dominante dell’endotelio corneale e della membrana di Descemet. Si presenta con edema corneale senza aumento del diametro corneale. L’esame dell’endotelio corneale è utile per la diagnosi.
Altre malattie costituenti ②
Cornea sclerotizzata : Invasione non infiammatoria e non progressiva di tessuto sclerale opaco nella cornea periferica, rendendo il confine tra cornea e sclera indistinto. Sono coinvolte mutazioni di FOXE3, PAX6, ecc.
Megalocornea : Malattia con diametro corneale ≥ 12,5 mm, spesso dovuta a mutazione del gene CHRDL1 (eredità recessiva legata all’X). La pressione intraoculare e le cellule endoteliali sono generalmente normali. La megalocornea congenita si distingue dal glaucoma congenito primario (PCG) per l’assenza di strie di Haab e di aumento dell’escavazione papillare.
I primi sintomi del glaucoma evolutivo sono segni di irritazione dovuti all’edema epiteliale corneale causato dall’aumento della pressione intraoculare. Nello specifico, si osservano lacrimazione senza secrezione, fotofobia e blefarospasmo. Se l’ipertensione oculare persiste, l’edema epiteliale corneale diventa grave, portando a opacità corneale. Inoltre, la capsula oculare (in particolare la giunzione corneo-congiuntivale) si stira, causando un aumento del diametro corneale (buftalmo) e un aumento della profondità della camera anteriore.
Quando la cornea viene stirata, la membrana di Descemet, meno elastica, si rompe (strie di Haab) e l’afflusso di umore acqueo nello stroma corneale aggrava rapidamente l’edema e l’opacità corneale. Le strie di Haab lasciano un’opacità permanente e sono causa di deficit visivo.
Segno
Valore normale / Riferimento
Diametro corneale del neonato
9,5–10,5 mm (a 1 anno 10,0–11,5 mm). > 12,0 mm subito dopo la nascita suggerisce PCG
Rapporto coppa/disco (C/D)
Nei lattanti, ≥ 0,3 suggerisce glaucoma. Una differenza interoculare ≥ 0,2 è anche indicativa di glaucoma
Segni associati ad anomalie dello sviluppo del segmento anteriore
Verificare i segni del segmento anteriore caratteristici di ciascuna malattia. L’embriotossone posteriore (spostamento anteriore/ispessimento della linea di Schwalbe) è associato ad ARS e sindrome di Alagille. L’ipoplasia dell’iride, la policoria e la deviazione dell’iride sono caratteristiche dell’ARS. L’opacità corneale centrale e le aderenze irido-corneali suggeriscono un’anomalia di Peters.
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
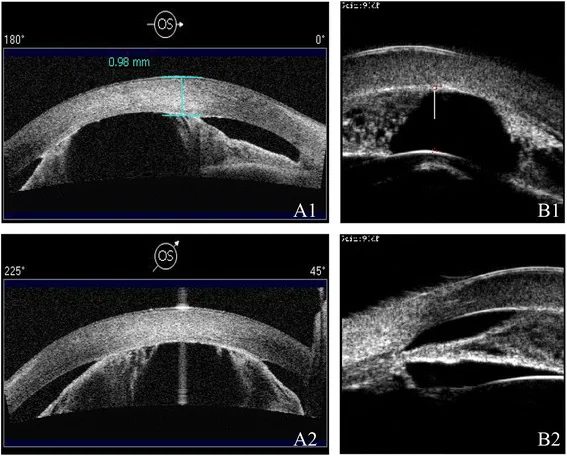
Le AS-OCT (A1/A2) e le UBM (B1/B2) mostrano una camera anteriore poco profonda e aderenze irido-corneali con diverse modalità. Si osserva un restringimento della camera anteriore e anomalie strutturali del segmento anteriore.
Secondo le linee guida per la cura del glaucoma (5a edizione) 6) e la classificazione internazionale del Childhood Glaucoma Research Network (CGRN) 7), il glaucoma pediatrico viene diagnosticato se sono soddisfatti almeno due dei seguenti cinque criteri.
Pressione intraoculare superiore a 21 mmHg
Progressione dell’aumento del rapporto cup/disc (C/D), aumento asimmetrico o assottigliamento del bordo neuroretinico
Segni corneali (strie di Haab, diametro corneale ≥11 mm nel neonato, ≥12 mm sotto 1 anno, ≥13 mm a qualsiasi età)
Miopeizzazione dovuta ad allungamento assiale oltre il normale sviluppo
Difetto del campo visivo riproducibile coerente con neuropatia ottica glaucomatosa
Nei bambini di età inferiore a 5 anni, spesso è necessario un esame in sedazione o anestesia generale. Sotto anestesia generale, la pressione intraoculare è più bassa rispetto allo stato di veglia, quindi è necessaria cautela nell’interpretazione delle misurazioni. L’esame dell’angolo viene eseguito con una lampada a fessura portatile e un gonioscopio diretto come la lente di Koeppe. In caso di opacità corneale che rende difficile l’osservazione con il gonioscopio, è utile l’ecobiomicroscopia (UBM).
La diagnosi del tipo di malattia viene effettuata mediante differenziazione basata sui reperti del segmento anteriore. Si verifica la presenza di anello embriotossico posteriore, anomalie dell’iride (ARS), cataratta (anomalia di Peters). Se si sospetta una causa ereditaria, si deve prendere in considerazione l’esame del glaucoma nei parenti e il test genetico.
QQual è il punto più importante da considerare nell'esame del glaucoma pediatrico?
A
Tutti i farmaci utilizzati in anestesia generale abbassano la pressione intraoculare (l’unica eccezione è la ketamina che può aumentarla leggermente). Pertanto, non si può escludere un glaucoma solo sulla base delle misurazioni in anestesia generale. È importante eseguire misurazioni in stato di veglia quando possibile e valutare complessivamente altri reperti oltre alla pressione intraoculare, come il diametro corneale, le strie di Haab e l’escavazione della papilla ottica.
Il glaucoma congenito precoce è fondamentalmente una malattia che richiede un trattamento chirurgico; la terapia farmacologica è considerata come abbassamento della pressione intraoculare a breve termine prima dell’intervento e come trattamento adiuvante postoperatorio. Poiché il paziente è un neonato, non è raro che sia necessario un reintervento; è importante spiegare bene questo ai genitori e ottenere la loro comprensione.
Come primo intervento si esegue una goniotomia o una trabeculotomia. Questi interventi agiscono direttamente sull’anomalia di sviluppo della via di deflusso dell’umor acqueo. In caso di insuccesso, si prendono in considerazione un intervento filtrante o uno shunt tubulare.
Anche dopo il controllo della pressione intraoculare, spesso è necessario un trattamento per l’ambliopia. L’anisometropia, l’astigmatismo irregolare, l’opacità corneale e le strie di Haab possono essere cause di ambliopia; pertanto, dopo l’intervento, è importante eseguire regolarmente esami dell’acuità visiva e della refrazione.
Nell’anomalia di Peters con grave opacità corneale, è indicato il trapianto di cornea (cheratoplastica perforante), ma nei casi gravi con aderenza cristallino-corneale, il rischio di fallimento del trapianto è elevato.
Nello sviluppo del segmento anteriore, l’epitelio corneale e il cristallino derivati dall’ectoderma superficiale, insieme all’epitelio pigmentato retinico derivato dal neuroectoderma, interagiscono con le cellule mesenchimali derivate dalla cresta neurale per formare una normale struttura del segmento anteriore. Le cellule della cresta neurale partecipano alla formazione dello stroma corneale, dell’endotelio corneale, del trabecolato e dello stroma irideo. Anomalie nella migrazione o differenziazione di queste cellule causano disgenesie del segmento anteriore.
Nell’ARS, FOXC1 e PITX2 interagiscono fisicamente e sono essenziali per la normale differenziazione delle cellule della cresta neurale. Mutazioni in entrambi i geni possono produrre fenotipi simili a causa di questa interazione.
QLe anomalie dello sviluppo del segmento anteriore possono essere diagnosticate in modo definitivo tramite test genetici?
A
Non in tutti i casi. Nel 40-75% dei casi la causa genetica non è ancora stata identificata. Tuttavia, i test genetici sono utili per la diagnosi differenziale (ad esempio, mutazione PAX6 nell’aniridia e valutazione del gene WT1 adiacente per escludere la sindrome WAGR), lo screening familiare e la consulenza genetica. In particolare nell’ARS, il 50-75% dei parenti può sviluppare glaucoma, pertanto si raccomandano test genetici e screening della pressione intraoculare e del glaucoma nei familiari.
QAnche i fattori ambientali sono un rischio per le anomalie dello sviluppo del segmento anteriore?
A
Uno studio caso-controllo coreano (582 casi vs 1.746 controlli) ha riportato che un aumento dell’esposizione materna al PM2,5 nei tre mesi precedenti il concepimento e durante il primo e secondo trimestre di gravidanza era associato a un aumento del rischio di ASD (aniridia, ipoplasia dell’iride, anomalia di Peters, ARS, PCG) nel bambino 8). Oltre ai fattori genetici, anche fattori ambientali come infezioni durante la gravidanza e sostanze teratogene possono interferire con il normale sviluppo del segmento anteriore.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.