Dị tật phát triển đoạn trước (anterior segment developmental anomalies; ASDA), còn được gọi là loạn sản đoạn trước (anterior segment dysgenesis; ASD), là thuật ngữ chung cho các rối loạn phát triển bẩm sinh xảy ra ở các cấu trúc đoạn trước như giác mạc, mống mắt, thể thủy tinh và góc tiền phòng1).
Các bệnh này rất đa dạng về kiểu hình và di truyền, nhưng tăng nhãn áp do bất thường phát triển đường dẫn lưu thủy dịch (bè củng giác mạc và ống Schlemm), tức glôcôm phát triển, là biến chứng quan trọng thường gặp 1,2).
QTại sao dị tật phát triển đoạn trước dễ bị glôcôm phát triển?
A
Các tế bào trung mô có nguồn gốc từ mào thần kinh tham gia vào sự phát triển của đoạn trước, và nhóm tế bào này cũng góp phần vào sự phát triển của bè củng giác mạc và ống Schlemm. Khi có bất thường trong hình thành đoạn trước, cấu trúc đường dẫn lưu thủy dịch thường bị ảnh hưởng đồng thời, dẫn đến tăng sức cản dẫn lưu và tăng nhãn áp. Do đó, glôcôm phát triển là biến chứng thường gặp của ASDA.
Hội chứng Axenfeld-Rieger (ARS) là bệnh di truyền trội trên nhiễm sắc thể thường, biểu hiện triệu chứng ở mắt và toàn thân. Dấu hiệu ở mắt bao gồm vòng phôi sau (đường Schwalbe dày lên và lệch về phía trước), giảm sản mống mắt, đa đồng tử, và lệch mống mắt. Có sự dính giữa đường Schwalbe nổi và mống mắt được gọi là bất thường Axenfeld; nếu kèm teo mống mắt hoặc nhu mô thì gọi là bất thường Rieger. Khi kèm bất thường xương hoặc răng, gọi là hội chứng Rieger. Giác mạc thường bình thường và cấu trúc nội mô bình thường, nhưng có thể bị đục thứ phát do tiếp xúc vật lý với các mô tồn dư. Đột biến gen yếu tố phiên mã PITX2 (nhiễm sắc thể 4) và FOXC1 (nhiễm sắc thể 6) là nguyên nhân, 50-75% bệnh nhân bị glôcôm1). Glôcôm có thể xuất hiện từ giai đoạn trẻ sơ sinh, nhưng hầu hết xuất hiện ở thời thơ ấu đến thanh niên. Kiểu hình khác nhau giữa đột biến FOXC1 và PITX2; báo cáo cho thấy bất thường giác mạc 50% ở đột biến FOXC1 và 16% ở đột biến PITX2 2).
Bệnh bẩm sinh đặc trưng bởi đục giác mạc trung tâm và khiếm khuyết nội mô và màng Descemet. 80% hai mắt, có thể kèm dính mống mắt-giác mạc hoặc thể thủy tinh-giác mạc. Đột biến PAX6, PITX2, CYP1B1 có liên quan. Hội chứng Peters plus có bất thường toàn thân như sứt môi, lùn, chậm phát triển tâm thần, do đột biến gen B3GLCT (di truyền lặn trên nhiễm sắc thể thường) 3). Trường hợp nặng cần ghép giác mạc, nhưng tiên lượng xấu nếu có dính thể thủy tinh-giác mạc3).
Bệnh di truyền lặn trên nhiễm sắc thể thường do bất thường phát triển của bè củng giác mạc. Đột biến phổ biến nhất là ở gen CYP1B1 (locus GLC3A, 2p21) 4). Trong một đoàn hệ trẻ em Nhật Bản bị glôcôm, đột biến CYP1B1 được phát hiện ở khoảng 20%, và đột biến FOXC1 cũng được ghi nhận 4). Ở Nhật Bản, tần suất khoảng 1/100.000, 75% hai mắt, 65% ở trẻ nam, và 80% xuất hiện trong năm đầu đời. Nhãn áp cao ở trẻ sơ sinh gây tăng đường kính giác mạc (mắt bò), phù và đục giác mạc, vỡ màng Descemet (vạch Haab).
Bệnh di truyền trội trên nhiễm sắc thể thường đặc trưng bởi giảm sản mống mắt, do đột biến gen PAX6 (nhiễm sắc thể 11) 5). Ngoài khiếm khuyết mống mắt một phần đến toàn bộ, có thể kèm lệch thể thủy tinh, đục giác mạc, và giảm thị lực do giảm sản hoàng điểm. Nếu kèm mất đoạn gen WT1 lân cận, có nguy cơ hội chứng WAGR (u Wilms, vô mống mắt, bất thường niệu dục, chậm phát triển tâm thần), cần đánh giá WT1. Trường hợp mất đoạn WT1, khuyến cáo siêu âm thận mỗi 3 tháng đến 8 tuổi và theo dõi với chuyên khoa ung thư nhi 5).
Các thành phần bệnh khác ①
Loạn dưỡng nội mô giác mạc bẩm sinh di truyền (CHED): Bệnh lặn nhiễm sắc thể thường, biểu hiện phù giác mạc đối xứng hai mắt từ khi sinh đến 1-2 tuổi. Nguyên nhân do đột biến gen SLC4A11, không kèm tăng nhãn áp.
Loạn dưỡng giác mạc đa dạng thể sau (PPCD): Bệnh trội nhiễm sắc thể thường ở nội mô giác mạc và màng Descemet. Biểu hiện phù giác mạc nhưng không tăng đường kính giác mạc. Xét nghiệm nội mô giác mạc hữu ích cho chẩn đoán.
Các bệnh thành phần khác ②
Giác mạc hóa củng mạc: Mô củng mạc đục xâm lấn vùng rìa giác mạc một cách không viêm và không tiến triển, làm ranh giới giác-củng mạc không rõ. Đột biến FOXE3, PAX6 có liên quan.
Giác mạc to (Megalocornea): Bệnh tăng đường kính giác mạc ≥12,5 mm, thường do đột biến CHRDL1 di truyền lặn liên kết X. Nhãn áp và tế bào nội mô thường bình thường. Giác mạc to bẩm sinh được phân biệt với PCG bởi không có vệt Haab hay lõm gai thị rộng.
Triệu chứng ban đầu của glôcôm phát triển là các triệu chứng kích thích do phù biểu mô giác mạc vì tăng nhãn áp. Cụ thể là chảy nước mắt không kèm ghèn, sợ ánh sáng, co thắt mi mắt. Nếu nhãn áp cao kéo dài, phù biểu mô trở nên nặng và gây đục giác mạc. Hơn nữa, vỏ nhãn cầu (đặc biệt vùng rìa giác mạc) bị giãn, dẫn đến tăng đường kính giác mạc (mắt bò) và tăng độ sâu tiền phòng.
Khi giác mạc giãn, màng Descemet kém đàn hồi bị rách (vệt Haab), gây tràn thủy dịch vào nhu mô giác mạc, làm phù và đục giác mạc nặng lên nhanh chóng. Vệt Haab để lại đục vĩnh viễn và gây suy giảm thị lực.
Dấu hiệu
Giá trị bình thường / Tiêu chuẩn
Đường kính giác mạc trẻ sơ sinh
9,5–10,5 mm (10,0–11,5 mm lúc 1 tuổi). Nếu >12,0 mm ngay sau sinh, nghi ngờ PCG
Tỷ lệ lõm gai thị (C/D)
Ở trẻ nhũ nhi, ≥0,3 nghi ngờ glôcôm. Chênh lệch ≥0,2 giữa hai mắt cũng gợi ý glôcôm
Các dấu hiệu liên quan đến bất thường phát triển đoạn trước
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
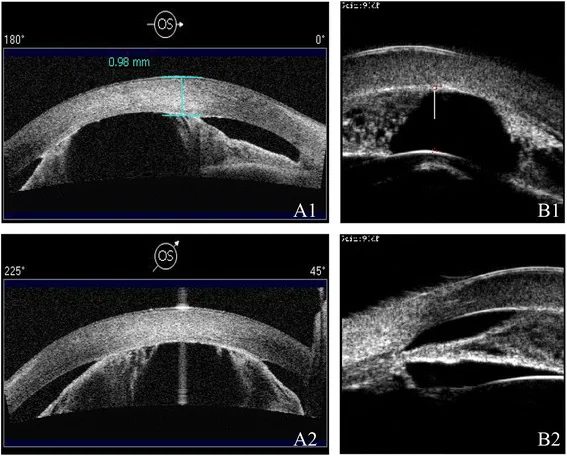
AS-OCT của A1/A2 và UBM của B1/B2 cho thấy tiền phòng nông và dính mống mắt-giác mạc bằng các phương thức khác nhau. Có thể thấy hẹp tiền phòng và bất thường cấu trúc đoạn trước.
Theo Hướng dẫn Thực hành Glôcôm (Phiên bản thứ 5) 6) và phân loại của Mạng lưới Nghiên cứu Glôcôm Trẻ em (CGRN) quốc tế 7), glôcôm trẻ em được chẩn đoán nếu đáp ứng hai hoặc nhiều hơn trong năm tiêu chí sau.
Nhãn áp vượt quá 21 mmHg
Tiến triển tăng tỷ lệ lõm gai thị (C/D), tăng bất đối xứng giữa hai mắt hoặc mỏng vành thần kinh
Dấu hiệu giác mạc (vạch Haab, đường kính giác mạc ≥11 mm ở trẻ sơ sinh, ≥12 mm ở trẻ dưới 1 tuổi, ≥13 mm ở mọi lứa tuổi)
Cận thị do dài trục nhãn cầu vượt quá phát triển bình thường
Ở trẻ dưới 5 tuổi, thường cần thăm khám dưới thuốc ngủ hoặc gây mê toàn thân. Dưới gây mê toàn thân, nhãn áp thấp hơn so với lúc tỉnh, do đó cần thận trọng khi giải thích kết quả đo. Khám góc tiền phòng được thực hiện bằng đèn khe cầm tay và kính soi góc trực tiếp như kính Koeppe. Nếu đục giác mạc gây khó quan sát bằng kính soi góc, kính hiển vi siêu âm sinh học (UBM) rất hữu ích.
Trong chẩn đoán thể bệnh, phân biệt dựa trên các dấu hiệu ở đoạn trước. Kiểm tra sự hiện diện của vòng phôi sau, bất thường mống mắt (ARS), hoặc đục thủy tinh thể (bất thường Peters). Nếu nghi ngờ di truyền, cân nhắc khám glôcôm cho người thân và xét nghiệm di truyền.
QĐiểm quan trọng nhất cần lưu ý khi thăm khám glôcôm trẻ em là gì?
A
Tất cả các thuốc dùng trong gây mê toàn thân đều làm giảm nhãn áp (ngoại lệ duy nhất là ketamine có thể làm tăng nhẹ). Do đó, không thể loại trừ glôcôm chỉ dựa trên đo nhãn áp dưới gây mê toàn thân. Cần thực hiện đo khi tỉnh càng nhiều càng tốt và đánh giá toàn diện các dấu hiệu khác như đường kính giác mạc, vạch Haab, và độ lõm đĩa thị.
Glôcôm bẩm sinh khởi phát sớm về cơ bản là bệnh cần điều trị phẫu thuật, và điều trị bằng thuốc được xem là hạ nhãn áp ngắn hạn trước phẫu thuật và điều trị hỗ trợ sau phẫu thuật. Vì bệnh nhân là trẻ sơ sinh, cần giải thích đầy đủ cho phụ huynh về khả năng phải phẫu thuật lại không hiếm gặp và đạt được sự đồng thuận.
Phẫu thuật đầu tiên thường là mở góc tiền phòng (goniotomy) hoặc mở bè củng mạc (trabeculotomy). Các phẫu thuật này can thiệp trực tiếp vào bất thường phát triển của đường dẫn lưu thủy dịch. Nếu không thành công, cân nhắc phẫu thuật lọc hoặc phẫu thuật ống dẫn lưu.
Sau khi kiểm soát nhãn áp, thường cần điều trị nhược thị. Chênh lệch khúc xạ, loạn thị không đều, đục giác mạc và vạch Haab có thể gây nhược thị, do đó cần khám thị lực và khúc xạ định kỳ sau phẫu thuật.
Trong bất thường Peters với đục giác mạc nặng, ghép giác mạc (ghép xuyên) được chỉ định, nhưng trong trường hợp nặng có dính thể thủy tinh-giác mạc, nguy cơ thất bại mảnh ghép cao.
Trong sự phát triển của đoạn trước, biểu mô giác mạc và thủy tinh thể có nguồn gốc từ ngoại bì bề mặt, và biểu mô sắc tố võng mạc có nguồn gốc từ ngoại bì thần kinh, tương tác với các tế bào trung mô có nguồn gốc từ mào thần kinh để hình thành cấu trúc đoạn trước bình thường. Tế bào mào thần kinh tham gia vào sự hình thành nhu mô giác mạc, nội mô giác mạc, bè củng mạc và nhu mô mống mắt. Sự bất thường trong di chuyển hoặc biệt hóa này gây ra các dị tật phát triển đoạn trước.
Trong hội chứng Axenfeld-Rieger, FOXC1 và PITX2 tương tác vật lý và đã được chứng minh là cần thiết cho sự biệt hóa bình thường của tế bào mào thần kinh. Đột biến ở một trong hai gen có thể tạo ra kiểu hình tương tự do sự tương tác này.
QCác bất thường phát triển đoạn trước có thể được chẩn đoán xác định bằng xét nghiệm di truyền không?
A
Không phải tất cả các trường hợp. Trong 40-75% trường hợp, nguyên nhân di truyền hiện vẫn chưa được xác định. Tuy nhiên, xét nghiệm di truyền hữu ích trong việc phân biệt các phân nhóm (ví dụ: đột biến PAX6 trong vô mống mắt và đánh giá gen WT1 lân cận để loại trừ hội chứng WAGR), sàng lọc trong gia đình và tư vấn di truyền. Đặc biệt trong hội chứng Axenfeld-Rieger, nơi 50-75% người thân có thể phát triển glôcôm, khuyến cáo xét nghiệm di truyền và kiểm tra nhãn áp, glôcôm cho các thành viên gia đình.
QCác yếu tố môi trường có phải là nguy cơ gây bất thường phát triển đoạn trước không?
A
Một nghiên cứu bệnh chứng tại Hàn Quốc (582 ca so với 1.746 đối chứng) báo cáo rằng việc tăng tiếp xúc của mẹ với PM2.5 trong 3 tháng trước khi thụ thai và trong tam cá nguyệt thứ nhất và thứ hai của thai kỳ có liên quan đến tăng nguy cơ ASD (vô mống mắt, thiểu sản mống mắt, bất thường Peters, ARS, PCG) ở trẻ8). Ngoài yếu tố di truyền, các yếu tố môi trường như nhiễm trùng khi mang thai và các chất gây quái thai cũng có thể cản trở sự phát triển bình thường của đoạn trước.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.