Las anomalías del desarrollo del segmento anterior (anterior segment developmental anomalies; ASDA), también llamadas disgenesia del segmento anterior (anterior segment dysgenesis; ASD), son un término general para los trastornos congénitos del desarrollo que afectan las estructuras del segmento anterior, como la córnea, el iris, el cristalino y el ángulo de la cámara anterior1).
Estas enfermedades son extremadamente diversas en fenotipo y genética, pero la elevación de la presión intraocular debida al desarrollo anormal de la vía de salida del humor acuoso (malla trabecular y canal de Schlemm), es decir, el glaucoma del desarrollo, es una complicación común e importante 1,2).
Q¿Por qué las anomalías del desarrollo del segmento anterior a menudo se complican con glaucoma del desarrollo?
A
El desarrollo del segmento anterior involucra células mesenquimales derivadas de la cresta neural, que también contribuyen al desarrollo de la malla trabecular y el canal de Schlemm. Cuando ocurren malformaciones del segmento anterior, la estructura de la vía de salida del humor acuoso a menudo se ve afectada simultáneamente, lo que lleva a un aumento de la resistencia a la salida del humor acuoso y a una elevación de la presión intraocular. Por lo tanto, el glaucoma del desarrollo es una complicación común de las ASDA.
El ARS es un trastorno autosómico dominante con manifestaciones oculares y sistémicas. Los hallazgos oculares incluyen embriotóxon posterior (línea de Schwalbe desplazada anteriormente y engrosada), hipoplasia del iris, policoria y corectopia. La anomalía de Axenfeld se refiere a una línea de Schwalbe prominente con adherencias al iris, mientras que la anomalía de Rieger incluye además atrofia del iris y del estroma. Cuando se acompaña de hallazgos sistémicos como anomalías óseas o dentales, se denomina síndrome de Rieger. La córnea generalmente es normal con estructura endotelial normal, pero puede ocurrir opacificación secundaria debido al contacto físico con restos. Las mutaciones en los genes del factor de transcripción PITX2 (cromosoma 4) y FOXC1 (cromosoma 6) son causantes, y el glaucoma complica el 50–75% de los casos 1). El glaucoma puede desarrollarse en la infancia, pero la mayoría de los casos ocurren en la niñez o la juventud. Las mutaciones de FOXC1 y PITX2 tienen diferentes fenotipos: se reportan anomalías corneales en el 50% de las mutaciones de FOXC1 y en el 16% de las mutaciones de PITX2 2).
Un trastorno congénito caracterizado por opacidad corneal central y defectos del endotelio corneal y la membrana de Descemet. El 80% son bilaterales y pueden asociarse con adherencias iridocorneales o adherencias cristalinocorneales. Las mutaciones en PAX6, PITX2 y CYP1B1 están implicadas. El síndrome de Peters plus incluye anomalías sistémicas como labio leporino, talla baja y discapacidad intelectual, causado por mutaciones en el gen B3GLCT (autosómico recesivo) 3). Los casos graves requieren trasplante de córnea, pero el pronóstico es malo cuando hay adherencias cristalinocorneales 3).
Un trastorno autosómico recesivo causado por una disgenesia de la malla trabecular. Las mutaciones en el gen CYP1B1 (locus GLC3A, 2p21) son las más comunes 4). En una cohorte japonesa de glaucoma pediátrico, se detectaron mutaciones de CYP1B1 en aproximadamente el 20%, y también se encontraron mutaciones de FOXC1 4). En Japón, la frecuencia es de aproximadamente 1 en 100,000; el 75% son bilaterales, el 65% ocurren en varones y el 80% se desarrollan dentro del primer año de vida. La presión intraocular elevada en lactantes causa aumento del diámetro corneal (buftalmos), edema/opacidad corneal y rotura de la membrana de Descemet (estrías de Haab).
Un trastorno autosómico dominante caracterizado principalmente por hipoplasia del iris, causado por mutaciones en el gen PAX6 (cromosoma 11) 5). Además de defectos parciales a completos del iris, puede asociarse con luxación del cristalino, opacidad corneal y deterioro visual debido a hipoplasia macular. La deleción del gen WT1 adyacente aumenta el riesgo de síndrome WAGR (tumor de Wilms, aniridia, anomalías genitourinarias, discapacidad intelectual), y es necesaria la evaluación de WT1. En casos con deleción de WT1, se recomienda ecografía renal cada 3 meses hasta los 8 años y seguimiento en oncología pediátrica 5).
Otras enfermedades constituyentes ①
Distrofia endotelial corneal hereditaria congénita (CHED): Enfermedad autosómica recesiva en la que aparece edema corneal bilateral simétrico desde el nacimiento hasta alrededor de 1-2 años de edad. Es causada por mutaciones en SLC4A11 y no se observa elevación de la presión intraocular.
Distrofia corneal polimorfa posterior (PPCD): Enfermedad autosómica dominante del endotelio corneal y la membrana de Descemet. Se presenta con edema corneal pero sin aumento del diámetro corneal. El examen del endotelio corneal es útil para el diagnóstico.
Otras enfermedades componentes ②
Córnea escleralizada: El tejido escleral opaco invade la córnea periférica de forma no inflamatoria y no progresiva, haciendo que el borde entre la córnea y la esclera sea indistinto. Se asocia con mutaciones en FOXE3, PAX6, etc.
Megalocórnea: Enfermedad en la que el diámetro corneal se agranda a 12.5 mm o más, a menudo por herencia recesiva ligada al X debida a mutaciones en el gen CHRDL1. La presión intraocular y las células endoteliales suelen ser normales. La megalocórnea congénita se diferencia del PCG por la ausencia de estrías de Haab y excavación papilar.
Los síntomas iniciales del glaucoma de desarrollo son síntomas irritativos del edema epitelial corneal debido a la elevación de la presión intraocular. Específicamente, se observan lagrimeo sin secreción, fotofobia y blefaroespasmo. Si la presión intraocular alta persiste, el edema epitelial corneal se vuelve severo, causando opacidad corneal. Además, la cubierta ocular (especialmente el limbo esclerocorneal) se estira, resultando en un aumento del diámetro corneal (buftalmos) y profundización de la cámara anterior.
Cuando la córnea se estira, se producen rupturas en la membrana de Descemet, menos elástica (estrías de Haab), y la entrada de humor acuoso en el estroma corneal empeora rápidamente el edema y la opacidad corneal. Las estrías de Haab dejan opacidad permanente y causan discapacidad visual.
Hallazgo
Valor normal/estándar
Diámetro corneal neonatal
9.5–10.5 mm (10.0–11.5 mm al año de edad). Si >12.0 mm inmediatamente después del nacimiento, sospechar PCG
Relación copa-disco del nervio óptico (relación C/D)
En lactantes, una relación ≥0.3 sugiere glaucoma. Una diferencia ≥0.2 entre ambos ojos también sugiere glaucoma
Hallazgos relacionados con la disgenesia del segmento anterior
Identifique los hallazgos característicos del segmento anterior para cada enfermedad. El embriotoxón posterior (desplazamiento anterior y engrosamiento de la línea de Schwalbe) se asocia con ARS y síndrome de Alagille. La hipoplasia del iris, la policoria y la corectopia del iris son características del ARS. La opacidad corneal central y las adherencias iridocorneales sugieren anomalía de Peters.
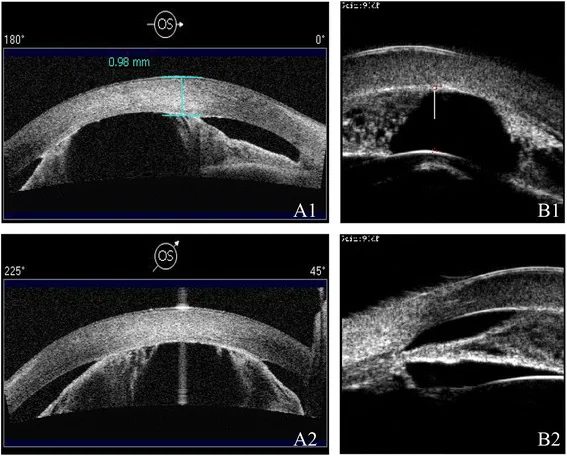
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
La AS-OCT (A1/A2) y la UBM (B1/B2) muestran cámara anterior poco profunda y adherencias iridocorneales en diferentes modalidades. Se observan el estrechamiento de la cámara anterior y anomalías de las estructuras del segmento anterior.
Según las Guías de Práctica Clínica para Glaucoma (5.ª edición) 6) y la clasificación internacional de la Red de Investigación de Glaucoma Infantil (CGRN) 7), se diagnostica glaucoma infantil cuando se cumplen dos o más de los siguientes cinco criterios.
Presión intraocular superior a 21 mmHg
Progresión del aumento de la relación copa-disco (relación C/D), aumento asimétrico o adelgazamiento del borde neuroretiniano
Hallazgos corneales (estrías de Haab, diámetro corneal ≥11 mm en recién nacidos, ≥12 mm en menores de 1 año, ≥13 mm en cualquier edad)
Miopía debida a elongación de la longitud axial más allá del desarrollo normal
Defecto del campo visual reproducible consistente con neuropatía óptica glaucomatosa
En niños menores de 5 años, a menudo es necesario realizar el examen bajo sedación o anestesia general. Bajo anestesia general, la presión intraocular es más baja que cuando está despierto, por lo que se debe tener precaución al interpretar las mediciones. La gonioscopia se realiza con una lámpara de hendidura portátil y un gonioscopio directo como la lente de Koeppe. Cuando la opacidad corneal dificulta la observación gonioscópica, la biomicroscopía ultrasónica (UBM) es útil.
En el diagnóstico del tipo de enfermedad, la diferenciación se basa en los hallazgos del segmento anterior. Verifique la presencia de embriotoxón posterior, anomalías del iris (ARS) y cataratas (anomalía de Peters). Si se sospecha una causa hereditaria, considere el examen de glaucoma en familiares y las pruebas genéticas.
Q¿Cuál es el punto más importante a tener en cuenta en el examen del glaucoma infantil?
A
Todos los fármacos utilizados en la anestesia general reducen la presión intraocular (la única excepción es la ketamina, que puede causar un ligero aumento). Por lo tanto, el glaucoma no puede descartarse basándose únicamente en las mediciones de la presión intraocular bajo anestesia general. Es importante realizar mediciones mientras el paciente está despierto siempre que sea posible y evaluar de manera integral los hallazgos distintos de la presión, como el diámetro corneal, las estrías de Haab y la excavación del disco óptico.
El glaucoma de desarrollo de inicio temprano es una enfermedad que fundamentalmente requiere tratamiento quirúrgico. La terapia médica se posiciona como una reducción de la presión intraocular a corto plazo antes de la cirugía y como tratamiento adyuvante después de la cirugía. Dado que el paciente es un lactante, es importante explicar completamente a los padres que la reoperación no es infrecuente y obtener su comprensión.
Como cirugía inicial, se realiza goniotomía o trabeculotomía. Estos procedimientos intervienen directamente en la anomalía del desarrollo de la vía de salida del humor acuoso. Si no tienen éxito, se considera la cirugía de filtración o la cirugía de derivación con tubo.
Incluso después de controlar la presión intraocular, a menudo es necesario el tratamiento de la ambliopía. La anisometropía, el astigmatismo irregular, la opacidad corneal y las estrías de Haab pueden causar ambliopía, por lo que se deben realizar exámenes regulares de agudeza visual y refracción después de la cirugía.
En la anomalía de Peters con opacidad corneal severa, está indicado el trasplante de córnea (queratoplastia penetrante), pero en casos graves con adherencia del cristalino a la córnea, el riesgo de fallo del injerto es alto.
En el desarrollo del segmento anterior, el epitelio corneal y el cristalino derivados del ectodermo superficial, y el epitelio pigmentario de la retina derivado del neuroectodermo interactúan con células mesenquimales derivadas de la cresta neural para formar estructuras normales del segmento anterior. Las células de la cresta neural participan en la formación del estroma corneal, el endotelio corneal, la malla trabecular y el estroma del iris. Las anomalías en su migración o diferenciación causan disgenesia del segmento anterior.
En ARS, FOXC1 y PITX2 interactúan físicamente y son esenciales para la diferenciación normal de las células de la cresta neural. Las mutaciones en cualquiera de los genes pueden producir fenotipos similares debido a esta interacción.
Q¿Se puede diagnosticar definitivamente la disgenesia del segmento anterior mediante pruebas genéticas?
A
No en todos los casos. En el 40–75% de los casos, actualmente no se identifica una causa genética. Sin embargo, las pruebas genéticas son útiles para diferenciar tipos de enfermedad (p. ej., mutación de PAX6 en aniridia vs. evaluación del gen WT1 adyacente para excluir el síndrome de WAGR), cribado familiar y asesoramiento genético. Particularmente en ARS, el 50–75% de los familiares pueden desarrollar glaucoma, por lo que se recomiendan pruebas genéticas y cribado de presión intraocular/glaucoma familiar.
Q¿Los factores ambientales también son un riesgo para las anomalías del desarrollo del segmento anterior?
A
Un estudio de casos y controles coreano (582 casos vs 1,746 controles) informó que el aumento de la exposición materna a PM2.5 durante los tres meses previos a la concepción y el primer y segundo trimestre del embarazo se asoció con un mayor riesgo de ASD (aniridia, hipoplasia del iris, anomalía de Peters, ARS, PCG) en los hijos 8). Además de los factores genéticos, los factores ambientales como las infecciones durante el embarazo y las sustancias teratogénicas también pueden alterar el desarrollo normal del segmento anterior.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.