Anomalien der Vorderabschnittsentwicklung (ASDA), auch als Vorderabschnittsdysgenesie (ASD) bezeichnet, sind ein Sammelbegriff für angeborene Entwicklungsstörungen der Strukturen des vorderen Augenabschnitts, einschließlich Hornhaut, Iris, Linse und Vorderkammerwinkel 1).
Diese Erkrankungen sind phänotypisch und genetisch äußerst vielfältig, aber eine gemeinsame wichtige Komplikation ist der Anstieg des Augeninnendrucks aufgrund einer Fehlentwicklung des Kammerwasserabflusstrakts (Trabekelwerk und Schlemm-Kanal), d. h. ein Entwicklungsglaukom1,2).
Zu den wichtigsten Erkrankungen, die zu den ASDA gehören, zählen der hintere Embryotoxon, das Axenfeld-Rieger-Syndrom (ARS), die Peters-Anomalie, das primäre kongenitale Glaukom (PCG), die Aniridie, die kongenitale hereditäre Endotheldystrophie (CHED), die posteriore polymorphe Hornhautdystrophie (PPCD), die Sklerokornea, die Megalokornea, das iridokorneale endotheliale (ICE) Syndrom usw.
QWarum sind Anomalien der Vorderabschnittsentwicklung häufig mit einem Entwicklungsglaukom assoziiert?
A
An der Entwicklung des vorderen Augenabschnitts sind mesenchymale Zellen aus der Neuralleiste beteiligt, die auch zur Entwicklung des Trabekelwerks und des Schlemm-Kanals beitragen. Wenn eine Fehlbildung des vorderen Augenabschnitts auftritt, ist die Struktur des Kammerwasserabflusstrakts oft ebenfalls beeinträchtigt, was zu einem erhöhten Abflusswiderstand und einem Anstieg des Augeninnendrucks führt. Daher ist das Entwicklungsglaukom eine häufige Komplikation der ASDA.
ARS ist eine autosomal-dominante Erkrankung mit okulären und systemischen Manifestationen. Zu den okulären Befunden gehören ein hinterer Embryotoxon (nach vorne verlagerte und verdickte Schwalbe-Linie), Iris-Hypoplasie, Polykorie und Iris-Abweichung. Eine prominente Schwalbe-Linie mit Verwachsungen an der Iris wird als Axenfeld-Anomalie bezeichnet, zusätzliche Atrophie der Iris oder des Stromas als Rieger-Anomalie. Bei Vorliegen von Skelett- oder Zahnanomalien spricht man vom Rieger-Syndrom. Die Hornhaut ist in der Regel normal, ebenso die Endothelstruktur, aber physikalischer Kontakt mit Restgewebe kann sekundär zu Trübungen führen. Mutationen der Transkriptionsfaktoren PITX2 (Chromosom 4) und FOXC1 (Chromosom 6) sind ursächlich, und in 50–75 % der Fälle tritt ein Glaukom auf 1). Das Glaukom kann bereits im Säuglingsalter auftreten, meist jedoch im Kindes- oder jungen Erwachsenenalter. Die Phänotypen unterscheiden sich: Bei FOXC1-Mutationen werden in 50 % Hornhautanomalien berichtet, bei PITX2-Mutationen in 16 % 2).
Eine angeborene Erkrankung, die durch eine zentrale Hornhauttrübung und das Fehlen von Hornhautendothel und Descemet-Membran gekennzeichnet ist. 80 % sind bilateral, und es können iridokorneale oder lentikulokorneale Verwachsungen auftreten. Mutationen von PAX6, PITX2 und CYP1B1 sind beteiligt. Das Peters-Plus-Syndrom umfasst systemische Anomalien wie Lippen-Kiefer-Gaumenspalte, Kleinwuchs und geistige Retardierung und wird durch Mutationen des B3GLCT-Gens (autosomal-rezessiv) verursacht 3). Schwere Fälle erfordern eine Hornhauttransplantation, aber die Prognose ist bei lentikulokornealen Verwachsungen schlecht 3).
Eine autosomal-rezessive Erkrankung, die durch eine Entwicklungsstörung des Trabekelwerks verursacht wird. Mutationen des CYP1B1-Gens (GLC3A-Locus, 2p21) sind am häufigsten 4). In einer japanischen Kohorte mit kindlichem Glaukom wurden CYP1B1-Mutationen bei etwa 20 % nachgewiesen, und auch FOXC1-Mutationen wurden berichtet 4). In Japan beträgt die Häufigkeit etwa 1 pro 100.000; 75 % sind bilateral, 65 % treten bei Jungen auf, und 80 % manifestieren sich innerhalb des ersten Lebensjahres. Erhöhter Augeninnendruck bei Säuglingen führt zu einer Vergrößerung des Hornhautdurchmessers (Buphthalmus), Hornhautödem und -trübung sowie Rissen der Descemet-Membran (Haab-Linien).
Eine autosomal-dominante Erkrankung, die hauptsächlich durch eine Iris-Hypoplasie gekennzeichnet ist und durch Mutationen des PAX6-Gens (Chromosom 11) verursacht wird 5). Neben einem partiellen oder vollständigen Fehlen der Iris können Linsenluxation, Hornhauttrübung und Sehbehinderung durch Makulahypoplasie auftreten. Eine Deletion des benachbarten WT1-Gens birgt das Risiko eines WAGR-Syndroms (Wilms-Tumor, Aniridie, genitourinäre Anomalien, geistige Retardierung), weshalb eine WT1-Bewertung erforderlich ist. Bei WT1-Deletion werden eine Nierenultraschalluntersuchung alle 3 Monate bis zum Alter von 8 Jahren und eine pädiatrisch-onkologische Nachsorge empfohlen 5).
Weitere konstituierende Erkrankungen ①
Kongenitale hereditäre Endotheldystrophie der Hornhaut (CHED) : Eine autosomal-rezessive Erkrankung, bei der von Geburt an bis zum Alter von etwa 1–2 Jahren ein beidseitiges, symmetrisches Hornhautödem auftritt. Ursache sind Mutationen im SLC4A11-Gen, ohne Erhöhung des Augeninnendrucks.
Polymorphe hintere Hornhautdystrophie (PPCD) : Eine autosomal-dominante Erkrankung des Hornhautendothels und der Descemet-Membran. Sie äußert sich in einem Hornhautödem ohne Vergrößerung des Hornhautdurchmessers. Die Untersuchung des Hornhautendothels ist für die Diagnose hilfreich.
Weitere konstituierende Erkrankungen ②
Skleralisierte Hornhaut : Nicht-entzündliches, nicht-progressives Eindringen von undurchsichtigem Skleragewebe in die periphere Hornhaut, wodurch die Grenze zwischen Hornhaut und Sklera undeutlich wird. Mutationen in FOXE3, PAX6 usw. sind beteiligt.
Megalokornea : Eine Erkrankung mit einem Hornhautdurchmesser von ≥ 12,5 mm, häufig verursacht durch eine Mutation im CHRDL1-Gen (X-chromosomal-rezessiv). Augeninnendruck und Endothelzellen sind in der Regel normal. Die angeborene Megalokornea wird vom primären kongenitalen Glaukom (PCG) dadurch unterschieden, dass keine Haab-Linien oder eine vergrößerte Papillenexkavation vorliegen.
Die ersten Symptome des Entwicklungsglaukoms sind Reizerscheinungen durch das Hornhautepithelödem infolge des erhöhten Augeninnendrucks. Konkret treten Tränenfluss ohne Sekret, Photophobie und Blepharospasmus auf. Bei anhaltendem hohen Druck wird das Hornhautepithelödem schwerwiegend und führt zu Hornhauttrübung. Darüber hinaus wird die Augenkapsel (insbesondere der korneokonjunktivale Übergang) gedehnt, was zu einer Vergrößerung des Hornhautdurchmessers (Buphthalmus) und einer Zunahme der Vorderkammertiefe führt.
Wenn die Hornhaut gedehnt wird, reißt die weniger elastische Descemet-Membran (Haab-Linien), und der Einstrom von Kammerwasser in das Hornhautstroma verschlimmert das Hornhautödem und die Trübung rasch. Haab-Linien hinterlassen eine dauerhafte Trübung und sind eine Ursache für Sehbehinderung.
Befund
Normalwert / Referenz
Hornhautdurchmesser des Neugeborenen
9,5–10,5 mm (mit 1 Jahr 10,0–11,5 mm). > 12,0 mm direkt nach der Geburt deutet auf PCG hin
Papillenexkavationsverhältnis (C/D)
Bei Säuglingen ≥ 0,3 deutet auf Glaukom hin. Ein Seitenunterschied ≥ 0,2 ist ebenfalls glaukomverdächtig
Befunde im Zusammenhang mit Vorderabschnittsfehlbildungen
Überprüfen Sie die für jede Erkrankung charakteristischen Vorderabschnittsbefunde. Der hintere Embryotoxon (anteriore Verlagerung/Verdickung der Schwalbe-Linie) tritt bei ARS und Alagille-Syndrom auf. Iris-Hypoplasie, Polykorie und Iris-Abweichung sind charakteristisch für ARS. Zentrale Hornhauttrübung und Iriskornealverwachsungen deuten auf eine Peters-Anomalie hin.
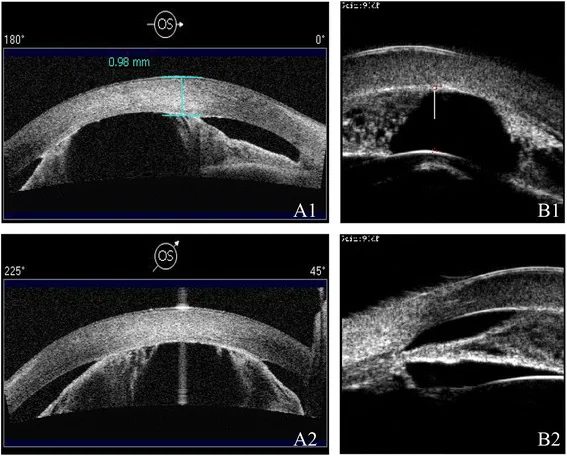
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
Die AS-OCT (A1/A2) und UBM (B1/B2) zeigen eine flache Vorderkammer und Iriskornealverwachsungen in verschiedenen Modalitäten. Die Verengung der Vorderkammer und die strukturellen Anomalien des Vorderabschnitts sind erkennbar.
Gemäß der Glaukom-Versorgungsleitlinie (5. Auflage) 6) und der internationalen Klassifikation des Childhood Glaucoma Research Network (CGRN) 7) wird ein kindliches Glaukom diagnostiziert, wenn mindestens zwei der folgenden fünf Kriterien erfüllt sind.
Augeninnendruck über 21 mmHg
Fortschreitende Vergrößerung des Cup/Disc-Verhältnisses (C/D), asymmetrische Vergrößerung oder Ausdünnung des neuroretinalen Randsaums
Hornhautbefunde (Haab-Linien, Hornhautdurchmesser ≥11 mm bei Neugeborenen, ≥12 mm unter 1 Jahr, ≥13 mm in jedem Alter)
Myopisierung durch über die normale Entwicklung hinausgehende Achsenlängenverlängerung
Reproduzierbarer Gesichtsfeldausfall, der mit einer glaukomatösen Optikusneuropathie vereinbar ist
Bei Kindern unter 5 Jahren ist die Untersuchung oft unter Sedierung oder Vollnarkose erforderlich. Unter Vollnarkose ist der Augeninnendruck niedriger als im Wachzustand, daher ist bei der Interpretation der Messwerte Vorsicht geboten. Die Kammerwinkeluntersuchung erfolgt mit einer Handspaltlampe und einem direkten Gonioskop wie der Koeppe-Linse. Bei Hornhauttrübung, die die Beobachtung mit dem Gonioskop erschwert, ist die Ultraschallbiomikroskopie (UBM) hilfreich.
Die Diagnose des Krankheitstyps erfolgt durch Differenzierung anhand der Vorderabschnittsbefunde. Es wird auf das Vorliegen eines hinteren Embryotoxons, von Irisanomalien (ARS), Katarakt (Peters-Anomalie) geachtet. Bei Verdacht auf eine erbliche Ursache sollten eine Glaukomuntersuchung bei Verwandten und eine genetische Testung in Betracht gezogen werden.
QWas ist der wichtigste Punkt bei der Untersuchung des kindlichen Glaukoms?
A
Alle in der Vollnarkose verwendeten Medikamente senken den Augeninnendruck (die einzige Ausnahme ist Ketamin, das ihn leicht erhöhen kann). Daher kann ein Glaukom allein aufgrund der Messungen unter Vollnarkose nicht ausgeschlossen werden. Es ist wichtig, nach Möglichkeit Messungen im Wachzustand durchzuführen und andere Befunde als den Augeninnendruck wie Hornhautdurchmesser, Haab-Linien und Exkavation der Papille insgesamt zu bewerten.
Das frühkindliche Entwicklungsglaukom ist grundsätzlich eine Erkrankung, die eine operative Behandlung erfordert; die medikamentöse Therapie wird als kurzfristige Drucksenkung bis zur Operation und als postoperative adjuvante Behandlung angesehen. Da der Patient ein Säugling ist, ist eine erneute Operation nicht selten erforderlich; es ist wichtig, dies den Eltern ausführlich zu erklären und ihr Verständnis zu gewinnen.
Als Ersteingriff werden eine Goniotomie oder eine Trabekulotomie durchgeführt. Diese Operationen greifen direkt in die Entwicklungsstörung des Kammerwasserabflusses ein. Bei Misserfolg werden eine filtrierende Operation oder eine Tube-Shunt-Operation in Betracht gezogen.
Auch nach Kontrolle des Augeninnendrucks ist häufig eine Amblyopiebehandlung erforderlich. Anisometropie, irregulärer Astigmatismus, Hornhauttrübung und Haab-Linien können Ursachen für eine Amblyopie sein; daher sollten nach der Operation regelmäßig Sehschärfe- und Refraktionsuntersuchungen durchgeführt werden.
Bei der Peters-Anomalie mit schwerer Hornhauttrübung ist eine Hornhauttransplantation (perforierende Keratoplastik) indiziert, aber bei schweren Fällen mit Linsen-Hornhaut-Verwachsung ist das Risiko eines Transplantatversagens hoch.
Bei der Entwicklung des vorderen Augenabschnitts interagieren das aus dem Oberflächenektoderm stammende Hornhautepithel und die Linse sowie das aus dem Neuroektoderm stammende retinale Pigmentepithel mit mesenchymalen Zellen aus der Neuralleiste, um eine normale Struktur des vorderen Augenabschnitts zu bilden. Neuralleistenzellen sind an der Bildung des Hornhautstromas, des Hornhautendothels, des Trabekelwerks und des Irisstromas beteiligt. Störungen der Migration oder Differenzierung dieser Zellen führen zu Dysgenesien des vorderen Augenabschnitts.
Bei ARS interagieren FOXC1 und PITX2 physikalisch und sind für die normale Differenzierung von Neuralleistenzellen unerlässlich. Mutationen in einem der Gene können aufgrund dieser Interaktion ähnliche Phänotypen hervorrufen.
QKönnen Entwicklungsanomalien des vorderen Augenabschnitts durch Gentests definitiv diagnostiziert werden?
A
Nicht in allen Fällen. Bei 40–75 % der Fälle wurde die genetische Ursache bisher nicht identifiziert. Gentests sind jedoch nützlich für die Differenzialdiagnose (z. B. PAX6-Mutation bei Aniridie und Bewertung des benachbarten WT1-Gens zum Ausschluss des WAGR-Syndroms), das familiäre Screening und die genetische Beratung. Insbesondere bei ARS können 50–75 % der Verwandten ein Glaukom entwickeln, daher werden Gentests sowie Augeninnendruck- und Glaukomuntersuchungen bei Familienmitgliedern empfohlen.
QSind Umweltfaktoren auch ein Risiko für Fehlbildungen des vorderen Augenabschnitts?
A
Eine koreanische Fall-Kontroll-Studie (582 Fälle vs. 1.746 Kontrollen) berichtete, dass eine erhöhte mütterliche PM2,5-Exposition in den drei Monaten vor der Empfängnis sowie im ersten und zweiten Schwangerschaftsdrittel mit einem erhöhten Risiko für ASD (Aniridie, Iris-Hypoplasie, Peters-Anomalie, ARS, PCG) beim Kind verbunden war 8). Neben genetischen Faktoren können auch Umweltfaktoren wie Infektionen während der Schwangerschaft und teratogene Substanzen die normale Entwicklung des vorderen Augenabschnitts beeinträchtigen.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.