ความผิดปกติของการพัฒนาส่วนหน้าของลูกตา (ASD A) เป็นคำรวมสำหรับความผิดปกติแต่กำเนิดของโครงสร้างส่วนหน้า เช่น กระจกตา ม่านตา ช่องหน้าลูกตา และเลนส์แก้วตา
รวมถึงกลุ่มอาการ Axenfeld-Rieger , ความผิดปกติของ Peters, โรคต้อหินแต่กำเนิด ปฐมภูมิ และภาวะไม่มีม่านตา
โรคต้อหิน จากการพัฒนาเนื่องจากความผิดปกติของทางระบายอารมณ์ขันน้ำเป็นภาวะแทรกซ้อนสำคัญที่พบบ่อย และอาจเกิดขึ้นได้ในผู้ป่วย 50-75%
ความผิดปกติของการย้ายที่และการแยกตัวของเซลล์นิวรัลคริสต์เป็นกลไกทางพยาธิวิทยาหลัก และมีรายงานยีนที่เกี่ยวข้องมากกว่า 50 ยีน รวมถึง PAX6, PITX2, FOXC1 และ CYP1B1
ความถี่ของโรคต้อหินแต่กำเนิด ปฐมภูมิในญี่ปุ่นประมาณ 1 ต่อ 100,000 คน และ 80% ปรากฏภายในปีแรกของชีวิต
ความผิดปกติของการพัฒนาส่วนหน้าของลูกตา (anterior segment developmental anomalies; ASD A) หรือที่เรียกว่า ภาวะเจริญผิดปกติของส่วนหน้า (anterior segment dysgenesis; ASD ) เป็นคำรวมสำหรับความผิดปกติแต่กำเนิดที่เกิดขึ้นในโครงสร้างส่วนหน้า เช่น กระจกตา ม่านตา เลนส์แก้วตา และมุมช่องหน้าลูกตา 1)
โรคเหล่านี้มีความหลากหลายมากทั้งในด้านฟีโนไทป์และพันธุกรรม แต่ความดันลูกตา สูงอันเนื่องมาจากความผิดปกติของการพัฒนาทางระบายอารมณ์ขันน้ำ (trabecular meshwork และ canal of Schlemm) หรือโรคต้อหิน จากการพัฒนา เป็นภาวะแทรกซ้อนสำคัญที่พบบ่อย 1,2)
โรคหลักที่รวมอยู่ใน ASD A ได้แก่: วงแหวนตัวอ่อนส่วนหลัง, กลุ่มอาการ Axenfeld-Rieger (ARS), ความผิดปกติของ Peters, โรคต้อหินแต่กำเนิด ปฐมภูมิ (PCG), ภาวะไม่มีม่านตา , โรคเสื่อมของเยื่อบุผนังกระจกตา ทางพันธุกรรมแต่กำเนิด (CHED ), โรคเสื่อมของกระจกตา ชนิดพหุสัณฐานส่วนหลัง (PPCD ), กระจกตา แข็ง, กระจกตา โต, และกลุ่มอาการเยื่อบุผนังกระจกตา กับม่านตา (ICE)
Q
ทำไมความผิดปกติของการพัฒนาส่วนหน้าของลูกตาจึงมีแนวโน้มที่จะเกิดโรคต้อหินจากการพัฒนา?
A
เซลล์มีเซนไคม์ที่มาจากนิวรัลคริสต์มีส่วนร่วมในการพัฒนาส่วนหน้าของลูกตา และกลุ่มเซลล์นี้ยังมีส่วนช่วยในการพัฒนา trabecular meshwork และ canal of Schlemm เมื่อเกิดความผิดปกติในการสร้างส่วนหน้า โครงสร้างของทางระบายอารมณ์ขันน้ำมักได้รับผลกระทบพร้อมกัน ทำให้ความต้านทานการระบายเพิ่มขึ้นและความดันลูกตา สูงขึ้น ดังนั้น โรคต้อหิน จากการพัฒนาจึงเป็นภาวะแทรกซ้อนที่พบบ่อยของ ASD A
กลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ (ARS) เป็นโรคถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่นที่แสดงอาการทางตาและระบบอื่นๆ ของร่างกาย การตรวจพบทางตา ได้แก่ วงแหวนตัวอ่อนด้านหลัง (เส้นชวาลเบที่เคลื่อนมาด้านหน้าและหนาตัวขึ้น), ม่านตา พร่อง (hypoplasia), รูม่านตา หลายรู (polycoria), และม่านตา เบี่ยงเบน การมีเส้นชวาลเบที่ยื่นเด่นและยึดติดกับม่านตา เรียกว่า ความผิดปกติแบบแอ็กเซนเฟลด์ (Axenfeld anomaly) ส่วนถ้ามีการฝ่อของม่านตา หรือสโตรมาร่วมด้วย เรียกว่า ความผิดปกติแบบรีเกอร์ (Rieger anomaly) ถ้ามีความผิดปกติของกระดูกหรือฟันร่วมด้วย เรียกว่า กลุ่มอาการรีเกอร์ (Rieger syndrome) กระจกตา ปกติและโครงสร้างเอนโดทีเลียมปกติ แต่อาจเกิดความขุ่นทุติยภูมิจากการสัมผัสทางกายภาพกับเศษเนื้อเยื่อ การกลายพันธุ์ของยีนถอดรหัส PITX2 (โครโมโซม 4) และ FOXC1 (โครโมโซม 6) เป็นสาเหตุ และ 50-75% มีโรคต้อหิน ร่วมด้วย 1) โรคต้อหิน อาจเกิดตั้งแต่ทารก แต่ส่วนใหญ่เกิดในวัยเด็กถึงวัยหนุ่มสาว ฟีโนไทป์แตกต่างกันระหว่างการกลายพันธุ์ FOXC1 และ PITX2 โดยรายงานความผิดปกติของกระจกตา 50% ใน FOXC1 และ 16% ใน PITX2 2)
โรคแต่กำเนิดที่มีลักษณะเด่นคือกระจกตา ขุ่นตรงกลางและข้อบกพร่องของเอนโดทีเลียมและเยื่อเดสเซเมต 80% เป็นสองตา และอาจมีการยึดติดระหว่างม่านตา กับกระจกตา หรือระหว่างเลนส์กับกระจกตา การกลายพันธุ์ของ PAX6, PITX2, CYP1B1 มีความเกี่ยวข้อง ในกลุ่มอาการปีเตอร์สพลัส (Peters plus syndrome) มีความผิดปกติของระบบอื่นๆ เช่น ปากแหว่ง, เตี้ย, พัฒนาการทางจิตช้า เกิดจากการกลายพันธุ์ของยีน B3GLCT (ถ่ายทอดแบบออโตโซมด้อย) 3) กรณีรุนแรงต้องปลูกถ่ายกระจกตา แต่พยากรณ์โรคไม่ดีถ้ามีการยึดติดระหว่างเลนส์กับกระจกตา 3)
โรคถ่ายทอดแบบออโตโซมด้อยจากความผิดปกติของพัฒนาการของ trabecular meshwork การกลายพันธุ์ที่พบบ่อยที่สุดคือในยีน CYP1B1 (ตำแหน่ง GLC3A, 2p21) 4) ในกลุ่มเด็กญี่ปุ่นที่เป็นต้อหิน พบการกลายพันธุ์ CYP1B1 ประมาณ 20% และยังพบการกลายพันธุ์ FOXC1 ด้วย 4) ในญี่ปุ่น ความถี่ประมาณ 1 ใน 100,000 คน 75% เป็นสองตา 65% ในเพศชาย และ 80% เกิดในปีแรกของชีวิต ความดันลูกตา สูงในทารกทำให้เส้นผ่านศูนย์กลางกระจกตา เพิ่มขึ้น (ตาวัว), กระจกตา บวมและขุ่น, และเยื่อเดสเซเมตแตก (เส้นฮาบ)
โรคถ่ายทอดแบบออโตโซมเด่นที่มีลักษณะเด่นคือม่านตา พร่อง เกิดจากการกลายพันธุ์ของยีน PAX6 (โครโมโซม 11) 5) นอกจากม่านตา บกพร่องบางส่วนถึงทั้งหมด อาจมีเลนส์เคลื่อน, กระจกตา ขุ่น, และการมองเห็น บกพร่องจาก macular hypoplasia ถ้ามีการขาดหายของยีน WT1 ที่อยู่ติดกัน จะเพิ่มความเสี่ยงของกลุ่มอาการ WAGR (วิลม์สทูเมอร์, ภาวะไม่มีม่านตา , ความผิดปกติของระบบสืบพันธุ์และทางเดินปัสสาวะ, ปัญญาอ่อน) จึงจำเป็นต้องประเมิน WT1 ในกรณีที่ขาด WT1 แนะนำให้อัลตราซาวนด์ไตทุก 3 เดือนจนถึงอายุ 8 ปี และติดตามผลกับแพทย์มะเร็งเด็ก 5)
องค์ประกอบอื่นของโรค ①
โรคกระจกตา เสื่อมชนิดเยื่อบุโพรงแต่กำเนิดที่ถ่ายทอดทางพันธุกรรม (CHED ) : โรคถ่ายทอดแบบออโตโซมัลรีเซสซีฟที่ทำให้เกิดอาการบวมน้ำที่กระจกตา ทั้งสองข้างแบบสมมาตรตั้งแต่แรกเกิดถึงอายุ 1-2 ปี เกิดจากการกลายพันธุ์ของยีน SLC4A11 โดยไม่มีความดันลูกตา สูง
โรคกระจกตา เสื่อมชนิดพหุสัณฐานส่วนหลัง (PPCD ) : โรคถ่ายทอดแบบออโตโซมัลโดมิแนนต์ของเยื่อบุโพรงกระจกตา และเยื่อเดสเซเมต์ ทำให้เกิดอาการบวมน้ำที่กระจกตา แต่ไม่มีเส้นผ่านศูนย์กลางกระจกตา เพิ่มขึ้น การตรวจเยื่อบุโพรงกระจกตา มีประโยชน์ในการวินิจฉัย
โรคประกอบอื่นๆ ②
กระจกตา กลายเป็นตาขาว (Sclerocornea)ตาขาว ทึบแสงรุกล้ำเข้ามาในกระจกตา ส่วนรอบแบบไม่มีการอักเสบและไม่ลุกลาม ทำให้ขอบระหว่างกระจกตา และตาขาว ไม่ชัดเจน การกลายพันธุ์ของยีน FOXE3, PAX6 มีความเกี่ยวข้อง
กระจกตา โต (Megalocornea)กระจกตา ≥12.5 มม. มักเกิดจากการกลายพันธุ์ของยีน CHRDL1 แบบถ่ายทอดทางโครโมโซม X แบบรีเซสซีฟ ความดันลูกตา และเซลล์เยื่อบุโพรงมักปกติ กระจกตาโตแต่กำเนิด แยกจาก PCG โดยไม่มีรอยแฮบหรือโพรงประสาทตาขยาย
อาการเริ่มแรกของโรคต้อหิน ในเด็กคืออาการระคายเคืองจากอาการบวมน้ำของเยื่อบุกระจกตา อันเนื่องมาจากความดันลูกตา สูง โดยเฉพาะมีน้ำตาไหลโดยไม่มีขี้ตา กลัวแสง และหนังตากระตุก หากความดันสูงต่อเนื่อง อาการบวมน้ำของเยื่อบุกระจกตา จะรุนแรงและทำให้กระจกตา ขุ่น นอกจากนี้ ผนังลูกตา (โดยเฉพาะบริเวณรอยต่อกระจกตา กับตาขาว ) จะยืดออก ทำให้เส้นผ่านศูนย์กลางกระจกตา เพิ่มขึ้น (ตาวัว) และความลึกของช่องหน้าม่านตา เพิ่มขึ้น
เมื่อกระจกตา ยืดออก เยื่อเดสเซเมต์ซึ่งยืดหยุ่นน้อยจะแตก (รอยแฮบ) ทำให้เกิดการไหลของอารมณ์ขันน้ำเข้าไปในเนื้อกระจกตา ส่งผลให้อาการบวมน้ำและความขุ่นของกระจกตา แย่ลงอย่างรวดเร็ว รอยแฮบทิ้งรอยขุ่นถาวรและทำให้การมองเห็น บกพร่อง
สิ่งที่ตรวจพบ ค่าปกติ/เกณฑ์อ้างอิง เส้นผ่านศูนย์กลางกระจกตา ทารกแรกเกิด 9.5–10.5 มม. (10.0–11.5 มม. เมื่ออายุ 1 ปี) หาก >12.0 มม. ทันทีหลังคลอด ให้สงสัย PCG อัตราส่วนร่องประสาทตา (C/D) ในทารก ค่า ≥0.3 สงสัยโรคต้อหิน ความแตกต่าง ≥0.2 ระหว่างสองตาก็บ่งชี้โรคต้อหิน
ยืนยันลักษณะเฉพาะของส่วนหน้าสำหรับแต่ละโรค วงแหวนตัวอ่อนส่วนหลัง (การเลื่อนไปข้างหน้าและหนาตัวของเส้นชวาลเบอ) สัมพันธ์กับกลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ และกลุ่มอาการอลาจิลล์ ภาวะม่านตา พร่อง (iris hypoplasia), ภาวะม่านตา หลายรู (polycoria) และการเบี่ยงเบนของม่านตา เป็นลักษณะเฉพาะของกลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ ภาวะกระจกตา ขุ่นกลางและการยึดติดระหว่างม่านตา กับกระจกตา บ่งชี้ถึงความผิดปกติของปีเตอร์ส
หากบุตรหลานของคุณมีอาการเช่น ตาดูใหญ่ น้ำตาไหลบ่อย หรือไวต่อแสง กรุณาพบจักษุแพทย์แต่เนิ่นๆ
โรคต้อหินแต่กำเนิด เป็นโรคที่ต้องได้รับการผ่าตัดรักษาตั้งแต่เนิ่นๆ การตรวจตาเป็นประจำมีความสำคัญอย่างยิ่งต่อการรักษาการทำงานของการมองเห็น อาจจำเป็นต้องให้คำปรึกษาทางพันธุกรรม กรุณาปรึกษาแพทย์ผู้รักษา
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PM
CI D: PMC4619091. License: CC BY.
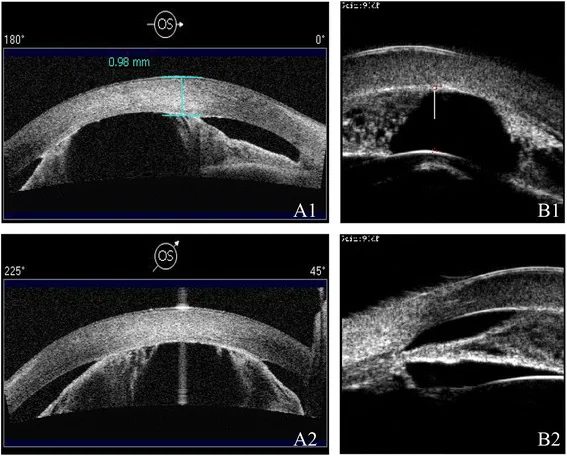
AS-OCT ของ A1/A2 และ
UBM ของ B1/B2 แสดงให้เห็น
ช่องหน้าม่านตาตื้น และการยึดติดระหว่าง
ม่านตา กับ
กระจกตา ด้วยวิธีการที่แตกต่างกัน จะเห็นการแคบลงของช่องหน้า
ม่านตา และความผิดปกติของโครงสร้างส่วนหน้า
ตามแนวทางปฏิบัติทางคลินิกสำหรับโรคต้อหิน (ฉบับที่ 5) 6) และการจำแนกประเภทของเครือข่ายวิจัยโรคต้อหิน ในเด็ก (CGRN) ระหว่างประเทศ 7) โรคต้อหิน ในเด็กได้รับการวินิจฉัยหากมีเกณฑ์อย่างน้อยสองในห้าข้อต่อไปนี้
ความดันลูกตา สูงกว่า 21 มิลลิเมตรปรอทการเพิ่มขึ้นของอัตราส่วนร่องประสาทตา (C/D) ที่ดำเนินไป, การเพิ่มขึ้นของความไม่สมมาตรระหว่างสองตา, หรือการบางลงของขอบประสาทตา
ลักษณะของกระจกตา (เส้นฮาบ, เส้นผ่านศูนย์กลางกระจกตา ≥11 มม. ในทารกแรกเกิด, ≥12 มม. ในเด็กอายุต่ำกว่า 1 ปี, ≥13 มม. ในทุกอายุ)
สายตาสั้น เนื่องจากการยืดตัวของความยาวแกนลูกตา ที่เกินกว่าการพัฒนาปกติความบกพร่องของลานสายตา ที่สามารถทำซ้ำได้ซึ่งสอดคล้องกับโรคเส้นประสาทตา จากต้อหิน
ในเด็กอายุต่ำกว่า 5 ปี มักจำเป็นต้องตรวจภายใต้การสะกดจิตหรือการดมยาสลบ ภายใต้การดมยาสลบ ความดันลูกตา จะต่ำกว่าขณะตื่น ดังนั้นจึงต้องระมัดระวังในการแปลผลการวัด การตรวจมุมลูกตาทำได้โดยใช้โคมไฟกรีดแบบมือถือและกล้องส่องมุมตาแบบตรง เช่น เลนส์ Koeppe หากกระจกตา ขุ่นทำให้การสังเกตด้วยกล้องส่องมุมตาทำได้ยาก กล้องจุลทรรศน์อัลตราซาวนด์ชีวภาพ (UBM ) จะมีประโยชน์
ในการวินิจฉัยชนิดของโรค จะแยกความแตกต่างจากลักษณะของส่วนหน้าของตา ตรวจสอบว่ามีวงแหวนตัวอ่อนด้านหลัง ความผิดปกติของม่านตา (ARS) หรือต้อกระจก (ความผิดปกติของปีเตอร์ส) หรือไม่ หากสงสัยว่ามีปัจจัยทางพันธุกรรม ให้พิจารณาตรวจต้อหิน ในญาติและการตรวจทางพันธุกรรม
Q
จุดที่สำคัญที่สุดที่ต้องระวังในการตรวจต้อหินในเด็กคืออะไร?
A
ยาทั้งหมดที่ใช้ในการดมยาสลบจะลดความดันลูกตา (ข้อยกเว้นเพียงอย่างเดียวคือคีตามีนซึ่งอาจเพิ่มขึ้นเล็กน้อย) ดังนั้นจึงไม่สามารถแยกแยะต้อหิน ได้จากการวัดความดันลูกตา ภายใต้การดมยาสลบเพียงอย่างเดียว สิ่งสำคัญคือต้องทำการวัดขณะตื่นให้มากที่สุดเท่าที่จะเป็นไปได้ และประเมินผลการตรวจอื่นๆ เช่น เส้นผ่านศูนย์กลางกระจกตา เส้น Haab และรอยบุ๋มของจานประสาทตา อย่างครอบคลุม
ต้อหิน แต่กำเนิดชนิดเริ่มต้นเร็วเป็นโรคที่ต้องรักษาด้วยการผ่าตัดเป็นหลัก และการรักษาด้วยยาถือเป็นการลดความดันลูกตา ในระยะสั้นก่อนการผ่าตัดและการรักษาเสริมหลังการผ่าตัด เนื่องจากผู้ป่วยเป็นทารก จึงสำคัญที่จะต้องอธิบายให้ผู้ปกครองเข้าใจว่าการผ่าตัดซ้ำไม่ใช่เรื่องแปลกและต้องได้รับความยินยอม
การผ่าตัดครั้งแรกมักเป็นการผ่าตัดเปิดมุมลูกตา (goniotomy) หรือการผ่าตัดเปิด trabeculum (trabeculotomy) การผ่าตัดเหล่านี้เป็นการแทรกแซงโดยตรงต่อความผิดปกติของพัฒนาการของทางระบายอารมณ์ขัน หากไม่สำเร็จ จะพิจารณาการผ่าตัดกรอง หรือการผ่าตัดท่อระบาย
หลังจากควบคุมความดันลูกตา ได้แล้ว มักจำเป็นต้องรักษาภาวะตาขี้เกียจ ภาวะสายตายาว หรือสั้นต่างกัน สายตาเอียง ไม่สม่ำเสมอ กระจกตา ขุ่น และเส้น Haab อาจทำให้เกิดภาวะตาขี้เกียจ ดังนั้นควรตรวจวัดสายตาและค่าสายตาเป็นระยะหลังการผ่าตัด
ในความผิดปกติของปีเตอร์สที่มีกระจกตา ขุ่นรุนแรง การปลูกถ่ายกระจกตา (การปลูกถ่ายกระจกตา ทั้งชั้น) เป็นข้อบ่งชี้ แต่ในกรณีรุนแรงที่มีการยึดติดของเลนส์กับกระจกตา ความเสี่ยงต่อความล้มเหลวของ graft สูง
ต้อหิน แต่กำเนิดยากที่จะควบคุมความดันลูกตา ในระยะยาวด้วยยาเพียงอย่างเดียว และหลักการคือการผ่าตัดตั้งแต่เนิ่นๆหากควบคุมความดันลูกตา ไม่เพียงพอ อาจเกิดการลุกลามของสายตาสั้น อย่างรวดเร็ว ควรวัดค่าสายตาและความยาวแกนลูกตา เป็นประจำเพื่อประเมินการลุกลามของโรคต้อหิน
ในเด็ก รอยบุ๋มของจานประสาทตา อาจเล็กลงหลังจากควบคุมความดันลูกตา ได้ ซึ่งเป็นตัวบ่งชี้การควบคุมความดันลูกตา ที่ดี
ในการพัฒนาส่วนหน้าของลูกตา เยื่อบุกระจกตา และเลนส์ที่มาจากเอ็กโทเดิร์มผิวหนัง และเยื่อบุผิวรงควัตถุจอประสาทตา ที่มาจากนิวรัลเอ็กโทเดิร์ม จะมีปฏิสัมพันธ์กับเซลล์มีเซนไคม์ที่มาจากนิวรัลคริสต์เพื่อสร้างโครงสร้างส่วนหน้าที่ปกติ เซลล์นิวรัลคริสต์มีส่วนร่วมในการสร้างสโตรมาของกระจกตา เอนโดทีเลียมของกระจกตา ทราเบคิวลาร์เมชเวิร์ก และสโตรมาของม่านตา ความผิดปกติในการย้ายที่หรือการแบ่งตัวนี้ทำให้เกิดความผิดปกติของการพัฒนาส่วนหน้า
มีการระบุยีนมากกว่า 50 ยีนที่เกี่ยวข้องกับ ASD A แต่ยังคงไม่ทราบสาเหตุทางพันธุกรรมใน 40-75% ของผู้ป่วย
ยีน โครโมโซม โรคที่เกี่ยวข้อง รูปแบบการถ่ายทอด PITX2 4q25 ARS, ปีเตอร์สอะนอมอลี ออโตโซมอลโดมิแนนต์ FOXC1 6p25 กลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์, ความผิดปกติของมุมม่านตา ถ่ายทอดแบบเด่นบนออโตโซม PAX6 11p13 ภาวะไม่มีม่านตา , ความผิดปกติของปีเตอร์ส ถ่ายทอดแบบเด่นบนออโตโซม CYP1B1 2p21 ต้อหิน แต่กำเนิดปฐมภูมิถ่ายทอดแบบด้อยบนออโตโซม
ในกลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ FOXC1 และ PITX2 มีปฏิสัมพันธ์ทางกายภาพ และได้รับการพิสูจน์ว่าจำเป็นต่อการเปลี่ยนแปลงที่ปกติของเซลล์ประสาทคริสต้า การกลายพันธุ์ในยีนใดยีนหนึ่งสามารถทำให้เกิดฟีโนไทป์ที่คล้ายกันได้เนื่องจากปฏิสัมพันธ์นี้
Q
ความผิดปกติของการพัฒนาส่วนหน้าของตาสามารถวินิจฉัยได้แน่ชัดด้วยการตรวจทางพันธุกรรมหรือไม่?
A
ไม่ใช่ในทุกกรณี ใน 40-75% ของกรณี ยังไม่มีการระบุสาเหตุทางพันธุกรรมในปัจจุบัน อย่างไรก็ตาม การตรวจทางพันธุกรรมมีประโยชน์ในการแยกแยะชนิดย่อย (เช่น การกลายพันธุ์ PAX6 ในภาวะไม่มีม่านตา และการประเมินยีน WT1 ที่อยู่ติดกันเพื่อแยกกลุ่มอาการ WAGR) การคัดกรองภายในครอบครัว และการให้คำปรึกษาทางพันธุกรรม โดยเฉพาะในกลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ ซึ่งญาติ 50-75% อาจเกิดต้อหิน แนะนำให้ตรวจทางพันธุกรรมและตรวจความดันลูกตา และต้อหิน ในสมาชิกครอบครัว
Q
ปัจจัยด้านสิ่งแวดล้อมเป็นความเสี่ยงต่อความผิดปกติของการพัฒนาส่วนหน้าของตาหรือไม่?
A
การศึกษาแบบ case-control ในเกาหลี (582 รายเทียบกับ 1,746 รายควบคุม) รายงานว่าการเพิ่มขึ้นของการสัมผัส PM2.5 ของมารดาในช่วง 3 เดือนก่อนตั้งครรภ์และในช่วงไตรมาสที่ 1 และ 2 ของการตั้งครรภ์สัมพันธ์กับความเสี่ยงที่เพิ่มขึ้นของ ASD (aniridia, iris hypoplasia, Peters anomaly, ARS, PCG) ในเด็ก8) นอกเหนือจากปัจจัยทางพันธุกรรมแล้ว ปัจจัยด้านสิ่งแวดล้อม เช่น การติดเชื้อระหว่างตั้งครรภ์และสารก่อวิรูป อาจขัดขวางการพัฒนาปกติของส่วนหน้าของตา
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCI D:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCI D:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCI D:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCI D:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCI D:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.