Ön segment gelişim anomalileri (ASDA), aynı zamanda ön segment disgenezisi (ASD) olarak da adlandırılır ve kornea, iris, lens, ön kamara açısı gibi ön segment yapılarında görülen konjenital gelişim bozukluklarını kapsayan bir kavramdır 1).
Bu hastalıklar fenotipik ve genetik olarak oldukça çeşitlidir, ancak aköz hümör drenaj yolunun (trabeküler ağ ve Schlemm kanalı) gelişimsel anomalisine bağlı göz içi basınç artışı, yani gelişimsel glokom, ortak ve önemli bir komplikasyondur 1,2).
QÖn segment gelişim anomalileri neden gelişimsel glokoma yatkınlık oluşturur?
A
Ön segmentin gelişiminde nöral krest kaynaklı mezenkimal hücreler rol oynar ve bu hücre grubu trabeküler ağ ve Schlemm kanalının gelişimine de katkıda bulunur. Ön segmentte gelişimsel bir anomali oluştuğunda, aköz hümör drenaj yolunun yapısı da sıklıkla etkilenir ve drenaj direnci artarak göz içi basıncı yükselir. Bu nedenle gelişimsel glokom, ASDA’nın yaygın bir komplikasyonudur.
ARS, göz ve sistemik bulgularla seyreden otozomal dominant bir hastalıktır. Göz bulguları arasında posterior embriyotokson (öne yer değiştirmiş ve kalınlaşmış Schwalbe çizgisi), iris hipoplazisi, polikori ve iris yer değiştirmesi yer alır. Belirgin Schwalbe çizgisi ve irise yapışıklık varsa Axenfeld anomalisi, ayrıca iris veya stromada atrofi varsa Rieger anomalisi olarak adlandırılır. Sistemik olarak kemik gelişim anomalileri ve diş anomalileri eşlik ediyorsa Rieger sendromu denir. Kornea genellikle normaldir ve endotel yapısı da normaldir, ancak kalıntılara fiziksel temas sekonder olarak bulanıklığa neden olabilir. PITX2 (kromozom 4) ve FOXC1 (kromozom 6) transkripsiyon faktör genlerindeki mutasyonlar nedendir ve vakaların %50-75’inde glokom eşlik eder 1). Glokom bazen bebeklik döneminde başlayabilir, ancak çoğunlukla çocukluk veya genç erişkinlikte ortaya çıkar. FOXC1 ve PITX2 mutasyonlarında fenotipler farklıdır; FOXC1 mutasyonunda kornea anomalisi %50, PITX2 mutasyonunda %16 olarak bildirilmiştir 2).
Santral kornea bulanıklığı ve kornea endoteli ile Descemet membranının yokluğu ile karakterize konjenital bir hastalıktır. %80’i bilateraldir ve iridokorneal veya lentikülokorneal yapışıklıklar eşlik edebilir. PAX6, PITX2 ve CYP1B1 mutasyonları ilişkilidir. Peters Plus sendromunda yarık dudak, kısa boy, zihinsel gerilik gibi sistemik anomaliler eşlik eder ve B3GLCT gen mutasyonu (otozomal resesif) nedenidir 3). Ağır vakalarda kornea nakli gerekir, ancak lentikülokorneal yapışıklık varsa prognoz kötüdür 3).
Trabeküler ağın gelişimsel anomalisine bağlı otozomal resesif bir hastalıktır. En sık CYP1B1 geni (GLC3A lokusu, 2p21) mutasyonu görülür 4). Japon çocuk glokom kohortunda CYP1B1 mutasyonu yaklaşık %20’de saptanmış ve FOXC1 mutasyonu da bildirilmiştir 4). Japonya’da sıklığı yaklaşık 100.000’de 1’dir, %75’i bilateral, %65’i erkek çocuklarda görülür ve %80’i yaşamın ilk yılı içinde ortaya çıkar. Bebeklerde yüksek göz içi basıncı kornea çapında artış (büyük göz), kornea ödemi ve bulanıklığı, Descemet membran yırtıkları (Haab çizgileri) oluşturur.
İris hipoplazisinin baskın olduğu otozomal dominant bir hastalıktır ve PAX6 geni (kromozom 11) mutasyonu nedenidir 5). İrisin kısmi veya tam yokluğuna ek olarak lens subluksasyonu, kornea bulanıklığı ve makula hipoplazisine bağlı görme azlığı eşlik edebilir. Komşu WT1 gen delesyonu varsa WAGR sendromu (Wilms tümörü, aniridi, genitoüriner anomaliler, zihinsel gerilik) riski vardır ve WT1 değerlendirmesi gereklidir. WT1 delesyonu olanlarda 8 yaşına kadar 3 ayda bir böbrek ultrasonu ve pediatrik onkoloji takibi önerilir 5).
Diğer yapısal hastalıklar ①
Konjenital Herediter Endotelyal Distrofi (CHED) : Doğumdan 1-2 yaşına kadar bilateral simetrik kornea ödemi ile ortaya çıkan otozomal resesif bir hastalıktır. SLC4A11 gen mutasyonu nedenidir ve göz içi basıncı yükselmez.
Posterior Polimorföz Kornea Distrofisi (PPCD) : Kornea endoteli ve Descemet membranının otozomal dominant bir hastalığıdır. Kornea ödemi ile seyreder ancak kornea çapında artış eşlik etmez. Kornea endotel incelemesi tanıda faydalıdır.
Diğer Bileşen Hastalıklar ②
Skleralize Kornea : Opak skleral dokunun periferik korneaya non-enflamatuar ve non-progresif olarak invaze olması ve kornea-sklera sınırının belirsizleşmesidir. FOXE3, PAX6 gibi gen mutasyonları ilişkilidir.
Megalokornea : Kornea çapının 12.5 mm veya daha fazla olduğu, çoğunlukla CHRDL1 gen mutasyonuna bağlı X’e bağlı resesif kalıtım gösteren bir hastalıktır. Göz içi basıncı ve endotel hücreleri genellikle normaldir. Konjenital megalokornea, Haab çizgileri ve optik disk çukurlaşması olmaması ile PCG’den ayırt edilir.
Gelişimsel glokomun ilk belirtisi, göz içi basıncı yükselmesine bağlı kornea epitel ödeminin irritasyon semptomlarıdır. Spesifik olarak, pürülan olmayan göz yaşarması, fotofobi ve blefarospazm görülür. Yüksek göz içi basıncı devam ederse kornea epitel ödemi şiddetlenir ve kornea bulanıklığı oluşur. Ayrıca göz küresi kapsülü (özellikle korneokonjonktival bileşke) gerilir, kornea çapında artış (büyük göz) ve ön kamara derinliğinde artış meydana gelir.
Kornea gerildiğinde, elastikiyeti düşük olan Descemet membranında yırtılmalar oluşur (Haab çizgileri) ve aköz hümörün kornea stromasına girmesiyle kornea ödemi ve bulanıklığı hızla kötüleşir. Haab çizgileri kalıcı bulanıklık bırakarak görme kaybına neden olur.
Bulgular
Normal Değer/Kriter
Yenidoğan kornea çapı
9.5-10.5 mm (1 yaşında 10.0-11.5 mm). Doğumdan hemen sonra 12.0 mm’yi aşarsa PCG’den şüphelenilir.
Optik disk çukurlaşma oranı (C/D)
Bebeklerde 0.3 ve üzeri glokomaçısından şüphelidir. İki göz arasında 0.2 ve üzeri fark da glokomu düşündürür.
Her hastalığa özgü ön segment bulguları değerlendirilir. Posterior embriyotokson (Schwalbe hattının öne yer değiştirmesi ve kalınlaşması) ARS ve Alagille sendromunda görülür. İris hipoplazisi, polikori ve iris deplasmanı ARS için karakteristiktir. Santral korneal opasite ve iridokorneal adezyonlar Peters anomalisini düşündürür.
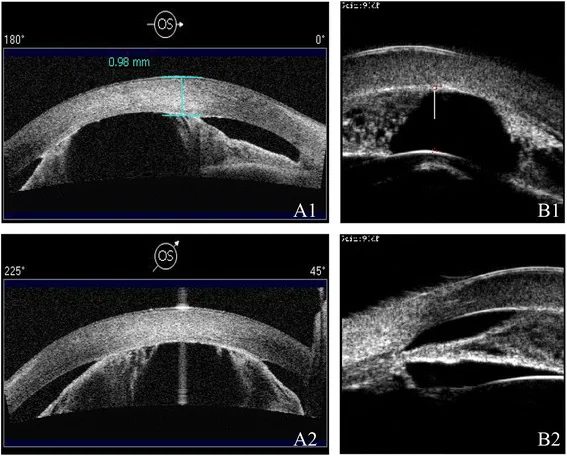
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
A1/A2’de AS-OCT ve B1/B2’de UBM ile sığ ön kamara ve iridokorneal adezyonlar farklı modalitelerde gösterilmiştir. Ön kamaranın daralması ve ön segment yapısal anomalileri görülmektedir.
Glokom Klinik Kılavuzu (5. baskı) 6) ve uluslararası Childhood Glaucoma Research Network (CGRN) sınıflamasına 7) göre, aşağıdaki beş kriterden en az ikisinin karşılanması durumunda pediatrik glokom tanısı konur.
Göz içi basıncının 21 mmHg’nin üzerinde olması
Optik disk çukurlaşma oranında (C/D oranı) ilerleyici artış, asimetrik artış veya rim incelmesi
Kornea bulguları (Haab çizgileri, yenidoğanda kornea çapı ≥11 mm, 1 yaş altında ≥12 mm, tüm yaşlarda ≥13 mm)
Normal gelişimin ötesinde aksiyel uzama nedeniyle miyopi gelişimi
Glokomatöz optik nöropati ile uyumlu, tekrarlanabilir görme alanı defekti
5 yaş altı çocuklarda genellikle sedasyon veya genel anestezi altında muayene gerekir. Genel anestezi altında göz içi basıncı uyanık haldekinden daha düşük olduğu için ölçüm değerlerinin yorumlanmasında dikkatli olunmalıdır. Açı muayenesi için el tipi yarık lamba ve Koeppe lensi gibi direkt goniyoskoplar kullanılır. Kornea bulanıklığı nedeniyle goniyoskopla gözlem zor olduğunda ultrasonik biyomikroskopi (UBM) yararlıdır.
Tip tanısında ön segment bulgularına göre ayırıcı tanı yapılır. Posterior embriyotokson, iris anomalisi (ARS), katarakt (Peters anomalisi) varlığı kontrol edilir. Kalıtsal olduğundan şüpheleniliyorsa akrabaların glokom muayenesi ve genetik test düşünülür.
QÇocukluk çağı glokomunda muayenede en dikkat edilmesi gereken nokta nedir?
A
Genel anestezide kullanılan tüm ilaçlar göz içi basıncını düşürür (tek istisna ketamindir, hafif yükseltebilir). Bu nedenle sadece genel anestezi altındaki göz içi basıncı ölçümü ile glokom dışlanamaz. Mümkün olduğunca uyanıkken ölçüm yapılmalı ve kornea çapı, Haab çizgileri, optik disk çukurluğu gibi göz içi basıncı dışındaki bulgular bütüncül olarak değerlendirilmelidir.
Erken başlangıçlı gelişimsel glokom temelde cerrahi tedavi gerektiren bir hastalıktır; ilaç tedavisi, cerrahiye kadar geçen sürede kısa süreli göz içi basıncı düşürülmesi ve cerrahi sonrası yardımcı tedavi olarak konumlandırılır. Hasta bebek veya küçük çocuk olduğu için tekrar cerrahi gerekmesinin nadir olmadığı ebeveynlere iyice açıklanmalı ve anlayışları sağlanmalıdır.
İlk cerrahi olarak goniyotomi veya trabekülotomi yapılır. Bu cerrahiler, aköz hümör çıkış yolundaki gelişimsel anomalilere doğrudan müdahale eden yöntemlerdir. Başarısız olunması durumunda filtrasyon cerrahisi veya tüp şant cerrahisi düşünülür.
Göz içi basıncı kontrol altına alındıktan sonra bile sıklıkla ambliyopi tedavisi gerekir. Refraktif anizometropi, düzensiz astigmatizma, kornea bulanıklığı ve Haab çizgileri ambliyopiye neden olabileceğinden, cerrahi sonrası düzenli olarak görme ve refraksiyon muayeneleri yapılmalıdır.
İleri kornea bulanıklığı olan Peters anomalisinde kornea nakli (tam kat kornea nakli) endikedir, ancak lens-kornea yapışıklığının eşlik ettiği ağır vakalarda greft yetmezliği riski yüksektir.
Ön segment gelişiminde, yüzey ektoderminden köken alan kornea epiteli ve lens ile nöral ektodermden köken alan retina pigment epiteli, nöral krest kaynaklı mezenkimal hücrelerle etkileşime girerek normal ön segment yapısını oluşturur. Nöral krest hücreleri, kornea stroması, kornea endoteli, trabeküler ağ ve iris stromasının oluşumunda rol oynar. Bu hücrelerin göç veya farklılaşma anormallikleri, ön segment gelişim anomalilerine yol açar.
ARS’de FOXC1 ve PITX2’nin fiziksel olarak etkileşime girdiği ve nöral krest hücrelerinin normal farklılaşması için gerekli olduğu gösterilmiştir. Her iki gendeki mutasyonun benzer fenotiplere yol açabilmesi bu etkileşimden kaynaklanmaktadır.
QÖn segment gelişim anomalileri genetik testle kesin olarak teşhis edilebilir mi?
A
Her vakada kesin teşhis mümkün değildir. Vakaların %40-75’inde şu anda genetik neden belirlenememektedir. Bununla birlikte, genetik test, hastalık tiplerinin ayırt edilmesinde (örneğin, aniridide PAX6 mutasyonu ve bitişik WT1 geninin değerlendirilmesiyle WAGR sendromunun dışlanması), aile içi taramada ve genetik danışmanlıkta faydalıdır. Özellikle ARS’de akrabaların %50-75’i glokom geliştirebileceğinden, genetik test ve ailede göz tansiyonu/glokom taraması önerilir.
QÇevresel faktörler de ön segment gelişim anomalileri için risk oluşturur mu?
A
Güney Kore’de yapılan bir vaka-kontrol çalışmasında (582 vaka vs 1746 kontrol), gebe kalmadan önceki 3 ay ve gebeliğin birinci ve ikinci trimesterlerinde annenin PM2.5 maruziyetindeki artışın, çocukta ASD (aniridi, iris hipoplazisi, Peters anomalisi, ARS, PCG) riskinde artışla ilişkili olduğu bildirilmiştir 8). Genetik faktörlerin yanı sıra, gebelik sırasındaki enfeksiyonlar ve teratojenik maddeler gibi çevresel faktörler de normal ön segment gelişimini bozabilir.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.