As anomalias do desenvolvimento do segmento anterior (anterior segment developmental anomalies; ASDA), também chamadas de disgenesias do segmento anterior (anterior segment dysgenesis; ASD), são um termo abrangente para distúrbios congênitos do desenvolvimento que ocorrem nas estruturas do segmento anterior, como córnea, íris, cristalino e ângulo da câmara anterior1).
Essas doenças são extremamente diversas fenotípica e geneticamente, mas o aumento da pressão intraocular devido a anormalidades no desenvolvimento da via de drenagem do humor aquoso (malha trabecular e canal de Schlemm), ou seja, o glaucoma de desenvolvimento, é uma complicação importante e comum 1,2).
QPor que as anomalias do desenvolvimento do segmento anterior são propensas a complicações de glaucoma de desenvolvimento?
A
Células mesenquimais derivadas da crista neural estão envolvidas no desenvolvimento do segmento anterior, e esse grupo celular também contribui para o desenvolvimento da malha trabecular e do canal de Schlemm. Quando ocorre uma anormalidade na formação do segmento anterior, a estrutura da via de drenagem do humor aquoso é frequentemente afetada simultaneamente, levando ao aumento da resistência ao fluxo e elevação da pressão intraocular. Portanto, o glaucoma de desenvolvimento é uma complicação comum da ASDA.
A síndrome de Axenfeld-Rieger (ARS) é uma doença autossômica dominante que apresenta sintomas oculares e sistêmicos. Os achados oculares incluem anel embrionário posterior (linha de Schwalbe deslocada anteriormente e espessada), hipoplasia da íris, policoria e desvio da íris. A presença de aderências entre a linha de Schwalbe proeminente e a íris é chamada de anomalia de Axenfeld; quando há também atrofia da íris ou do estroma, é chamada de anomalia de Rieger. Quando acompanhada de anormalidades ósseas ou dentárias, é chamada de síndrome de Rieger. A córnea geralmente é normal e a estrutura endotelial é normal, mas pode ocorrer opacificação secundária devido ao contato físico com resíduos. Mutações nos genes dos fatores de transcrição PITX2 (cromossomo 4) e FOXC1 (cromossomo 6) causam a doença, e 50-75% desenvolvem glaucoma1). O glaucoma pode surgir na infância, mas na maioria dos casos aparece na infância ou na juventude. O fenótipo difere entre mutações FOXC1 e PITX2; relata-se anormalidade corneana em 50% das mutações FOXC1 e 16% das mutações PITX2 2).
Doença congênita caracterizada por opacidade central da córnea e defeito do endotélio e membrana de Descemet. 80% são bilaterais, podendo haver aderências iridocorneanas ou lenticulocorneanas. Mutações em PAX6, PITX2 e CYP1B1 estão associadas. Na síndrome de Peters plus, há anormalidades sistêmicas como lábio leporino, baixa estatura e retardo mental, causada por mutações no gene B3GLCT (herança autossômica recessiva) 3). Casos graves requerem transplante de córnea, mas o prognóstico é ruim quando há aderência lenticulocorneana 3).
Doença autossômica recessiva causada por anormalidade do desenvolvimento da malha trabecular. As mutações mais comuns são no gene CYP1B1 (locus GLC3A, 2p21) 4). Em uma coorte japonesa de glaucoma infantil, mutações CYP1B1 foram detectadas em cerca de 20%, e mutações FOXC1 também foram relatadas 4). No Japão, a frequência é de cerca de 1 em 100.000, 75% bilaterais, 65% em meninos e 80% aparecem no primeiro ano de vida. A pressão intraocular elevada em lactentes causa aumento do diâmetro da córnea (buftalmia), edema e opacidade da córnea, e rupturas da membrana de Descemet (linhas de Haab).
Doença autossômica dominante caracterizada por hipoplasia da íris, causada por mutações no gene PAX6 (cromossomo 11) 5). Além de defeito parcial a total da íris, pode haver luxação do cristalino, opacidade da córnea e deficiência visual devido à hipoplasia macular. Se houver deleção do gene WT1 adjacente, há risco de síndrome WAGR (tumor de Wilms, aniridia, anormalidades urogenitais, retardo mental), sendo necessária avaliação do WT1. Em casos de deleção do WT1, recomenda-se ultrassonografia renal a cada 3 meses até os 8 anos de idade e acompanhamento com oncologia pediátrica 5).
Outros componentes da doença ①
Distrofia endotelial corneana hereditária congênita (CHED): Doença autossômica recessiva caracterizada por edema corneano simétrico bilateral desde o nascimento até 1-2 anos de idade. Causada por mutação no SLC4A11, sem aumento da pressão intraocular.
Distrofia corneana polimorfa posterior (PPCD): Doença autossômica dominante do endotélio corneano e membrana de Descemet. Apresenta edema corneano, mas sem aumento do diâmetro corneano. O exame do endotélio corneano é útil para o diagnóstico.
Outras Doenças Componentes ②
Córneaesclerocórnea: Tecido escleral opaco invade a córnea periférica de forma não inflamatória e não progressiva, tornando a borda córneo-escleral indistinta. Mutações em FOXE3, PAX6 estão associadas.
Megalocórnea: Aumento do diâmetro corneano ≥12,5 mm, frequentemente por mutação no CHRDL1 com herança recessiva ligada ao X. A pressão intraocular e as células endoteliais geralmente são normais. A megalocórnea congênita é diferenciada do PCG pela ausência de estrias de Haab ou escavação papilar aumentada.
Os sintomas iniciais do glaucoma de desenvolvimento são sintomas de irritação devido ao edema do epitélio corneano causado pelo aumento da pressão intraocular. Especificamente, observam-se lacrimejamento sem secreção, fotofobia e blefaroespasmo. Se a hipertensão persistir, o edema epitelial torna-se grave e leva à opacidade corneana. Além disso, a cápsula ocular (especialmente a região limbar) se distende, resultando em aumento do diâmetro corneano (olho de boi) e aumento da profundidade da câmara anterior.
Quando a córnea se distende, a membrana de Descemet, menos elástica, rompe-se (estrias de Haab), causando influxo de humor aquoso no estroma corneano, agravando rapidamente o edema e a opacidade. As estrias de Haab deixam opacidade permanente e causam deficiência visual.
Achado
Valor Normal / Referência
Diâmetro corneano do recém-nascido
9,5–10,5 mm (10,0–11,5 mm com 1 ano). Se >12,0 mm logo após o nascimento, suspeitar de PCG
Relação escavação/disco óptico (C/D)
Em lactentes, ≥0,3 é suspeito para glaucoma. Diferença ≥0,2 entre os olhos também sugere glaucoma
Achados relacionados a anomalias do desenvolvimento do segmento anterior
Confirmar os achados característicos do segmento anterior para cada doença. O anel embrionário posterior (deslocamento anterior e espessamento da linha de Schwalbe) está associado à síndrome de Axenfeld-Rieger e à síndrome de Alagille. Hipoplasia da íris, policoria e desvio da íris são características da síndrome de Axenfeld-Rieger. Opacidade corneana central e aderência íris-córnea sugerem anomalia de Peters.
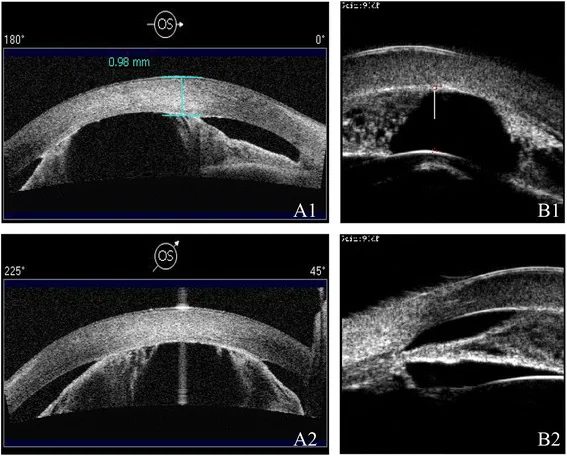
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
AS-OCT de A1/A2 e UBM de B1/B2 mostram câmara anterior rasa e aderência íris-córnea em diferentes modalidades. Observa-se estreitamento da câmara anterior e anormalidades estruturais do segmento anterior.
De acordo com as Diretrizes de Prática Clínica para Glaucoma (5ª edição) 6) e a classificação internacional da Childhood Glaucoma Research Network (CGRN) 7), o glaucoma pediátrico é diagnosticado se dois ou mais dos cinco critérios a seguir forem atendidos.
Pressão intraocular superior a 21 mmHg
Progressão do aumento da relação escavação/disco óptico (C/D), aumento da assimetria entre os olhos ou afinamento do anel neurorretiniano
Achados corneanos (linhas de Haab, diâmetro corneano ≥11 mm em neonatos, ≥12 mm em menores de 1 ano, ≥13 mm em todas as idades)
Miopia devido ao alongamento do comprimento axial além do desenvolvimento normal
Defeito de campo visual reproduzível consistente com neuropatia óptica glaucomatosa
Em crianças menores de 5 anos, os exames frequentemente precisam ser realizados sob hipnose ou anestesia geral. Sob anestesia geral, a pressão intraocular é mais baixa do que quando acordado, portanto, a interpretação das medidas requer cuidado. O exame do ângulo é realizado com lâmpada de fenda portátil e gonioscópio direto, como a lente de Koeppe. Se a opacidade da córnea dificultar a observação com gonioscópio, o microscópio ultrassônico de biomicroscopia (UBM) é útil.
No diagnóstico do tipo, a diferenciação é feita com base nos achados do segmento anterior. Verifique a presença de anel embrionário posterior, anomalia da íris (ARS) ou catarata (anomalia de Peters). Se houver suspeita de hereditariedade, considere o exame de glaucoma em parentes e testes genéticos.
QQual é o ponto mais importante a ser observado no exame de glaucoma infantil?
A
Todos os medicamentos usados na anestesia geral reduzem a pressão intraocular (a única exceção é a cetamina, que pode aumentá-la ligeiramente). Portanto, o glaucoma não pode ser descartado apenas com base nas medidas de pressão intraocular sob anestesia geral. É importante realizar medidas quando acordado sempre que possível e avaliar de forma abrangente outros achados, como diâmetro da córnea, linhas de Haab e escavação do disco óptico.
O glaucoma congênito de início precoce é uma doença que requer tratamento cirúrgico basicamente, e a terapia medicamentosa é posicionada como redução da pressão intraocular a curto prazo até a cirurgia e terapia adjuvante pós-operatória. Como o paciente é um bebê, é importante explicar aos pais que a necessidade de re-operação não é incomum e obter sua compreensão.
Como primeira cirurgia, são realizadas goniotomia ou trabeculotomia. Essas cirurgias intervêm diretamente na anomalia do desenvolvimento da via de drenagem do humor aquoso. Se não forem bem-sucedidas, considera-se cirurgia de filtração ou cirurgia de tubo de derivação.
Após o controle da pressão intraocular, muitas vezes é necessário tratamento para ambliopia. Anisometropia, astigmatismo irregular, opacidade da córnea e linhas de Haab podem causar ambliopia, portanto, exames de visão e refração devem ser realizados periodicamente após a cirurgia.
Na anomalia de Peters com opacidade grave da córnea, o transplante de córnea (ceratoplastia penetrante) é indicado, mas em casos graves com aderência córneo-lenticular, o risco de falha do enxerto é alto.
No desenvolvimento do segmento anterior, o epitélio corneano e o cristalino derivados do ectoderma superficial, e o epitélio pigmentar da retina derivado do ectoderma neural, interagem com células mesenquimais derivadas da crista neural para formar a estrutura normal do segmento anterior. As células da crista neural participam da formação do estroma corneano, endotélio corneano, malha trabecular e estroma da íris. Anormalidades na migração ou diferenciação dessas células causam anomalias de desenvolvimento do segmento anterior.
Na síndrome de Axenfeld-Rieger, FOXC1 e PITX2 interagem fisicamente, e foi demonstrado que são essenciais para a diferenciação normal das células da crista neural. Mutações em qualquer um dos genes podem produzir fenótipos semelhantes devido a essa interação.
QAs anomalias do desenvolvimento do segmento anterior podem ser diagnosticadas definitivamente por teste genético?
A
Não em todos os casos. Em 40-75% dos casos, a causa genética ainda não foi identificada atualmente. No entanto, o teste genético é útil para diferenciar subtipos (por exemplo, mutação PAX6 na aniridia e avaliação do gene WT1 adjacente para excluir a síndrome de WAGR), triagem intrafamiliar e aconselhamento genético. Especialmente na síndrome de Axenfeld-Rieger, onde 50-75% dos parentes podem desenvolver glaucoma, recomenda-se teste genético e exame de pressão ocular e glaucoma nos familiares.
QFatores ambientais também são um risco para anormalidades do desenvolvimento do segmento anterior?
A
Um estudo caso-controle coreano (582 casos vs 1.746 controles) relatou que o aumento da exposição materna a PM2.5 durante os 3 meses anteriores à concepção e no primeiro e segundo trimestres da gravidez está associado a um maior risco de ASD (aniridia, hipoplasia da íris, anomalia de Peters, ARS, PCG) na criança8). Além de fatores genéticos, fatores ambientais como infecções durante a gravidez e substâncias teratogênicas também podem prejudicar o desenvolvimento normal do segmento anterior.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.