Anterior segment developmental anomalies (ASDA), also called anterior segment dysgenesis (ASD), is a general term for congenital developmental disorders affecting anterior segment structures such as the cornea, iris, lens, and anterior chamber angle 1).
These diseases are extremely diverse in phenotype and genetics, but elevated intraocular pressure due to abnormal development of the aqueous humor outflow pathway (trabecular meshwork and Schlemm’s canal), i.e., developmental glaucoma, is a common and important complication 1,2).
QWhy do anterior segment developmental anomalies often complicate developmental glaucoma?
A
The development of the anterior segment involves neural crest-derived mesenchymal cells, which also contribute to the development of the trabecular meshwork and Schlemm’s canal. When anterior segment malformations occur, the structure of the aqueous humor outflow pathway is often simultaneously impaired, leading to increased outflow resistance and elevated intraocular pressure. Therefore, developmental glaucoma is a common complication of ASDA.
ARS is an autosomal dominant disorder with ocular and systemic manifestations. Ocular findings include posterior embryotoxon (anteriorly displaced and thickened Schwalbe’s line), iris hypoplasia, polycoria, and corectopia. Axenfeld anomaly refers to prominent Schwalbe’s line with iris adhesions, while Rieger anomaly additionally includes iris and stromal atrophy. When accompanied by systemic findings such as skeletal or dental abnormalities, it is called Rieger syndrome. The cornea is generally normal with normal endothelial structure, but secondary opacification may occur due to physical contact with remnants. Mutations in the transcription factor genes PITX2 (chromosome 4) and FOXC1 (chromosome 6) are causative, and glaucoma complicates 50–75% of cases 1). Glaucoma may develop in infancy, but most cases occur in childhood to young adulthood. FOXC1 and PITX2 mutations have different phenotypes: corneal abnormalities are reported in 50% of FOXC1 mutations and 16% of PITX2 mutations 2).
A congenital disorder characterized by central corneal opacity and defects of the corneal endothelium and Descemet’s membrane. 80% are bilateral, and may be associated with iridocorneal adhesions or lens-corneal adhesions. Mutations in PAX6, PITX2, and CYP1B1 are implicated. Peters plus syndrome includes systemic abnormalities such as cleft lip, short stature, and intellectual disability, caused by mutations in the B3GLCT gene (autosomal recessive) 3). Severe cases require corneal transplantation, but prognosis is poor when lens-corneal adhesions are present 3).
An autosomal recessive disorder caused by trabecular meshwork dysgenesis. Mutations in the CYP1B1 gene (GLC3A locus, 2p21) are most common 4). In a Japanese pediatric glaucoma cohort, CYP1B1 mutations were detected in about 20%, and FOXC1 mutations were also found 4). In Japan, the frequency is about 1 in 100,000; 75% are bilateral, 65% occur in males, and 80% develop within the first year of life. Elevated intraocular pressure in infants causes increased corneal diameter (buphthalmos), corneal edema/opacity, and Descemet’s membrane rupture (Haab’s striae).
An autosomal dominant disorder primarily characterized by iris hypoplasia, caused by mutations in the PAX6 gene (chromosome 11) 5). In addition to partial to complete iris defects, it may be associated with lens dislocation, corneal opacity, and visual impairment due to macular hypoplasia. Deletion of the adjacent WT1 gene increases the risk of WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, intellectual disability), and WT1 evaluation is necessary. In cases with WT1 deletion, renal ultrasound every 3 months until age 8 and pediatric oncology follow-up are recommended 5).
Other constituent diseases ①
Congenital hereditary endothelial dystrophy (CHED): An autosomal recessive disease in which bilateral symmetric corneal edema appears from birth to around 1–2 years of age. It is caused by mutations in SLC4A11, and intraocular pressure is not elevated.
Posterior polymorphous corneal dystrophy (PPCD): An autosomal dominant disease of the corneal endothelium and Descemet’s membrane. It presents with corneal edema but without an increase in corneal diameter. Corneal endothelial examination is useful for diagnosis.
Other component diseases ②
Sclerocornea: Opaque scleral tissue non-inflammatorily and non-progressively invades the peripheral cornea, blurring the border between cornea and sclera. Mutations in FOXE3, PAX6, etc. are associated.
Megalocornea: A disease in which the corneal diameter enlarges to 12.5 mm or more, often due to X-linked recessive inheritance caused by mutations in the CHRDL1 gene. Intraocular pressure and endothelial cells are usually normal. Congenital megalocornea is differentiated from PCG by the absence of Haab’s striae and optic disc cupping.
The initial symptoms of developmental glaucoma are irritative symptoms of corneal epithelial edema due to elevated intraocular pressure. Specifically, tearing without discharge, photophobia, and blepharospasm are observed. If high intraocular pressure persists, corneal epithelial edema becomes severe, leading to corneal opacity. Furthermore, the ocular coat (especially the corneoscleral limbus) is stretched, resulting in an increase in corneal diameter (buphthalmos) and deepening of the anterior chamber.
When the cornea is stretched, ruptures occur in the less elastic Descemet’s membrane (Haab’s striae), and the influx of aqueous humor into the corneal stroma rapidly worsens corneal edema and opacity. Haab’s striae leave permanent opacity and cause visual impairment.
Finding
Normal value/standard
Neonatal corneal diameter
9.5–10.5 mm (10.0–11.5 mm at 1 year of age). If >12.0 mm immediately after birth, suspect PCG
Optic disc cup-to-disc ratio (C/D ratio)
In infants, a ratio ≥0.3 suggests glaucoma. A difference of ≥0.2 between eyes also suggests glaucoma
Identify characteristic anterior segment findings for each disease. Posterior embryotoxon (anterior displacement and thickening of Schwalbe’s line) is associated with ARS and Alagille syndrome. Iris hypoplasia, polycoria, and iris corectopia are characteristic of ARS. Central corneal opacity and iridocorneal adhesions suggest Peters anomaly.
Ni W, Wang W, Sun S, et al. A novel histopathologic finding in the Descemet’s membrane of a patient with Peters Anomaly: a case-report and literature review. BMC Ophthalmol. 2015 Oct 23;15:139. Figure 2. PMCID: PMC4619091. License: CC BY.
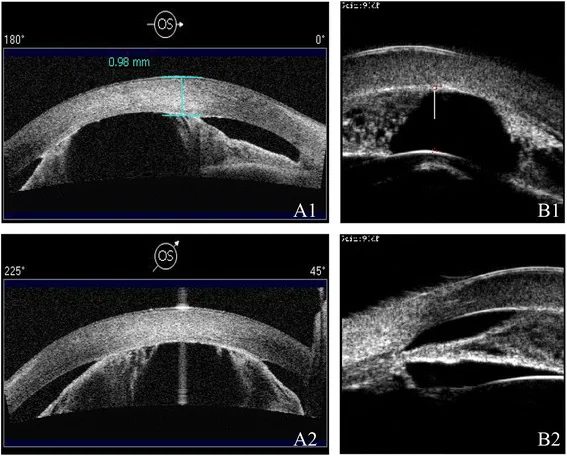
AS-OCT (A1/A2) and UBM (B1/B2) show shallow anterior chamber and iridocorneal adhesions using different modalities. Narrowing of the anterior chamber and abnormalities of anterior segment structures are visible.
According to the Glaucoma Clinical Practice Guidelines (5th edition) 6) and the international Childhood Glaucoma Research Network (CGRN) classification 7), childhood glaucoma is diagnosed when two or more of the following five criteria are met.
Intraocular pressure exceeds 21 mmHg
Progression of increased cup-to-disc ratio (C/D ratio), asymmetric increase, or thinning of the neuroretinal rim
Corneal findings (Haab striae, corneal diameter ≥11 mm in newborns, ≥12 mm in infants under 1 year, ≥13 mm at any age)
Myopia due to axial length elongation beyond normal development
Reproducible visual field defect consistent with glaucomatous optic neuropathy
In children under 5 years of age, examination under sedation or general anesthesia is often necessary. Under general anesthesia, intraocular pressure is lower than when awake, so caution is needed in interpreting measurements. Gonioscopy is performed using a handheld slit lamp and a direct gonioscope such as a Koeppe lens. When corneal opacity makes gonioscopic observation difficult, ultrasound biomicroscopy (UBM) is useful.
In the diagnosis of the disease type, differentiation is made based on anterior segment findings. Check for the presence of posterior embryotoxon, iris abnormalities (ARS), and cataract (Peters anomaly). If a hereditary cause is suspected, consider glaucoma examination of relatives and genetic testing.
QWhat is the most important point to note in the examination of childhood glaucoma?
A
All drugs used in general anesthesia lower intraocular pressure (the only exception is ketamine, which may cause a slight increase). Therefore, glaucoma cannot be ruled out based solely on intraocular pressure measurements under general anesthesia. It is important to perform measurements while awake whenever possible and to comprehensively evaluate non-pressure findings such as corneal diameter, Haab striae, and optic disc cupping.
Early-onset developmental glaucoma is a disease that fundamentally requires surgical treatment. Medical therapy is positioned as a short-term intraocular pressure reduction until surgery and as adjunctive treatment after surgery. Since the patient is an infant, it is important to fully explain to the parents that reoperation is not uncommon and to obtain their understanding.
As initial surgery, goniotomy or trabeculotomy is performed. These procedures directly intervene in the developmental abnormality of the aqueous outflow pathway. If unsuccessful, filtering surgery or tube shunt surgery is considered.
Even after intraocular pressure is controlled, amblyopia treatment is often necessary. Anisometropia, irregular astigmatism, corneal opacity, and Haab striae can cause amblyopia, so visual acuity and refraction should be checked regularly after surgery.
In Peters anomaly with severe corneal opacity, corneal transplantation (penetrating keratoplasty) is indicated, but in severe cases with lens-corneal adhesion, the risk of graft failure is high.
In anterior segment development, the corneal epithelium and lens derived from surface ectoderm, and the retinal pigment epithelium derived from neuroectoderm interact with neural crest-derived mesenchymal cells to form normal anterior segment structures. Neural crest cells are involved in the formation of the corneal stroma, corneal endothelium, trabecular meshwork, and iris stroma. Abnormalities in their migration or differentiation lead to anterior segment dysgenesis.
In ARS, FOXC1 and PITX2 physically interact and are essential for normal differentiation of neural crest cells. Mutations in either gene can produce similar phenotypes due to this interaction.
QCan anterior segment dysgenesis be definitively diagnosed by genetic testing?
A
Not in all cases. In 40–75% of cases, a genetic cause is not currently identified. However, genetic testing is useful for differentiating disease types (e.g., PAX6 mutation in aniridia vs. evaluation of adjacent WT1 gene to exclude WAGR syndrome), family screening, and genetic counseling. Particularly in ARS, 50–75% of relatives may develop glaucoma, so genetic testing and family intraocular pressure/glaucoma screening are recommended.
QAre environmental factors also a risk for anterior segment developmental anomalies?
A
A Korean case-control study (582 cases vs 1,746 controls) reported that increased maternal PM2.5 exposure during the three months before conception and the first and second trimesters was associated with an increased risk of ASD (aniridia, iris hypoplasia, Peters anomaly, ARS, PCG) in children 8). In addition to genetic factors, environmental factors such as infections during pregnancy and teratogenic substances may also disrupt normal anterior segment development.
Michels K, Bohnsack BL. Ophthalmological Manifestations of Axenfeld-Rieger Syndrome: Current Perspectives. Clinical ophthalmology (Auckland, N.Z.). 2023;17:819-828. doi:10.2147/OPTH.S379853. PMID:36926528; PMCID:PMC10013571.
Prem Senthil M, Knight LSW, Taranath D, Mackey DA, Ruddle JB, Chiang MY, et al. Comparison of Anterior Segment Abnormalities in Individuals With FOXC1 and PITX2 Variants. Cornea. 2022;41(8):1009-1015. doi:10.1097/ICO.0000000000003020. PMID:35354164; PMCID:PMC9390227.
Khasnavis A, Fernandes M. Peters anomaly: An overview. Taiwan journal of ophthalmology. 2023;13(4):434-442. doi:10.4103/tjo.TJO-D-23-00065. PMID:38249502; PMCID:PMC10798386.
Fuse N, Kimura M, Shimizu A, Koshiba S, Hamanaka T, Nakamura M, et al. Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals. Japanese journal of ophthalmology. 2024;68(6):688-701. doi:10.1007/s10384-024-01103-0. PMID:39158757; PMCID:PMC11607050.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
Kiuchi Y, Inoue T, Shoji N, et al. The Japan Glaucoma Society guidelines for glaucoma 5th edition. Jpn J Ophthalmol. 2023;67(2):189-254. doi:10.1007/s10384-022-00970-9. PMID:36780040.
Thau A, Lloyd M, Freedman S, et al. New classification system for pediatric glaucoma: implications for clinical care and a research registry. Curr Opin Ophthalmol. 2018;29(5):385-394. doi:10.1097/ICU.0000000000000516. PMID:30096087.
Choe S, Lee KS, Ha A, et al. Association of maternal exposure to fine particulate matter during pregnancy with anterior segment dysgenesis risk: a matched case-control study. J Clin Med. 2025;14(9):3003. doi:10.3390/jcm14093003.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.