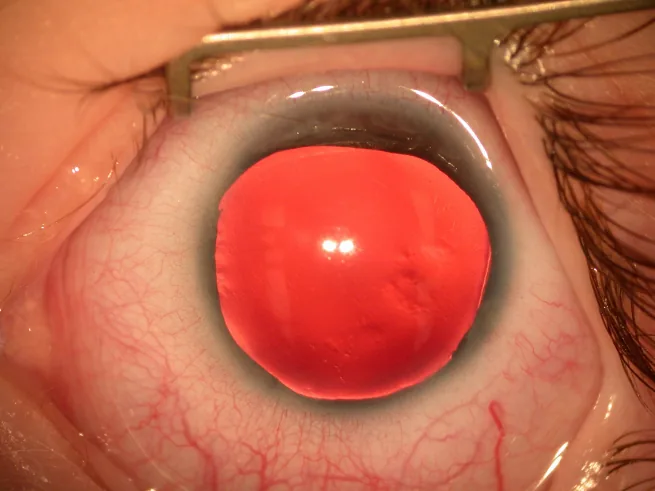
Aniridi, konjenital bir yatkınlık nedeniyle irisin tam veya kısmi yokluğu ile karakterize bir durumdur. “Aniridi” olarak adlandırılsa da, açının en periferik kısmında iris kökü sıklıkla kalır.
2017 yılında Sağlık, Çalışma ve Refah Bakanlığı’nın Nadir Hastalıklar Yasası kapsamında belirlenmiş nadir bir hastalık olarak tanınmıştır 1). Belirlenmiş nadir hastalık olarak teşhis edilen ve şiddet sınıflamasında derece III veya üzeri olarak değerlendirilen hastalar, tıbbi maliyet desteğine hak kazanır ve gelire göre belirlenmiş bir kişisel ödeme üst sınırı uygulanır 2).
İsveç ve Norveç’te yapılan epidemiyolojik çalışmalarda, prevalansın yaklaşık 90.000’de 1 olduğu bildirilmiştir3). PAX6 gen mutasyonu olan 43 hastanın detaylı oftalmolojik değerlendirmesinde, iris displazisinin derecesinin mutasyon tipine göre değiştiği gösterilmiştir3).
QAniridi kalıtsal mıdır?
A
Vakaların yaklaşık 2/3’ü otozomal dominant kalıtım gösterir ve etkilenen ebeveynden çocuğa %50 oranında geçme olasılığı vardır. Kalan 1/3’ü sporadiktir ve aile öyküsü yoktur. Sporadik vakalarda Wilms tümörü (böbrek tümörü) ile birlikte görülen WAGR sendromu riski nedeniyle PAX6 ve WT1 genlerinin genetik testi önerilir.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Ön segmentin yarık lamba fotoğrafında, iris neredeyse tamamen yok ve çevrede sadece çok ince bir iris kalıntısı görülüyor. Aniridinin tipik klinik bulgusunu doğrudan göstermekte olup, ana belirtiler ve klinik bulguların açıklamasına uygundur.
İrisin yokluğu veya eksikliği nedeniyle pupil işlev göremez ve göze giren ışık miktarı ayarlanamaz. Bu nedenle şiddetli fotofobi görülür. Ayrıca maküler hipoplaziye bağlı fiksasyon bozukluğu, erken bebeklik döneminden itibaren görülen horizontal nistagmusun ana nedeni olur.
Fotofobi: İrisin ışık ayarlayamaması → şiddetli parlama
Nistagmus (horizontal nistagmus): Maküler hipoplaziye bağlı fiksasyon bozukluğu. Erken bebeklik döneminden itibaren ortaya çıkar
Görme azlığı: Maküler hipoplazi, katarakt, glokom ve limbal kök hücre yetmezliğinin (LSCD) birleşik etkisi
PAX6 geni, göz dokularının yanı sıra merkezi sinir sistemi, pankreas Langerhans adacıkları ve koku epitelinde de ifade edilir. Bu dokuların hipoplazisi çeşitli göz dışı komplikasyonlara yol açabilir 1).
Korpus kallozum agenezisi, epilepsi, yüksek kortikal fonksiyon bozukluğu
Görme prognozu genellikle kötüdür ve çoğunlukla 0,1 civarındadır. Ancak foveal hipoplazinin derecesine ve komplikasyonların varlığına bağlı olarak 0,1 ile 0,7 arasında değişen bireysel farklılıklar vardır. Foveal hipoplazi için şu anda etkili bir tedavi yoktur ve en büyük görme kısıtlayıcı faktördür. Uygun refraktif düzeltme ve az görme rehabilitasyonu ile yaşam kalitesi iyileştirilebilir.
Aniridinin nedeni, 11. kromozomun kısa kolunda (11p13) yer alan PAX6 geninin bir alelinin fonksiyon kaybıdır (haplo-yetersizlik). Fonksiyonel gen dozunun yarıya inmesi sonucu oluşur. Her iki alelin anormal olması durumunda embriyonik ölümle sonuçlandığı düşünülmektedir 1).
PAX6, embriyonik dönemde organ farklılaşmasını düzenleyen transkripsiyon faktörlerinin ana kontrol genidir ve çeşitli transkripsiyon faktörlerini yönetir. PAX6 anormalliği, gözün tamamını etkileyen çeşitli konjenital anomalilere (aniridi, Peters anomalisi, foveal hipoplazi vb.) neden olur.
Gen mutasyon tipleri çoğunlukla nonsense, frameshift gibi prematüre trunkasyon kodon (PTC) tipi mutasyonlardır ve missense mutasyonlar da bildirilmiştir 1). İzole aniridinin sekans analizinde vakaların yaklaşık %85’inde PAX6 mutasyonu saptanır 2).
WAGR sendromu (sporadik vakalarda dikkat edilmesi gerekenler)
PAX6 geni, tümör baskılayıcı gen WT1 ile 11p13 kromozomu üzerinde komşudur. Sporadik vakalarda, komşu gen delesyonu nedeniyle Wilms tümörü, aniridi, genitoüriner anomaliler ve zihinsel gerilikten oluşan WAGR sendromu görülebilir 3). Sporadik vakaların yaklaşık %30’unda Wilms tümörü 5 yaşına kadar erken bilateral olarak ortaya çıkar.
PAX6 mutasyonu pozitif ve WT1 delesyonu yok → WAGR sendromu olasılığının olmadığı tahmin edilebilir 2)
Genetik test, DNA sekanslama + MLPA/CMA ile genomik yapısal anormalliklerin saptanmasının kombinasyonu ile yapılır 2)
Sporadik vakalarda WAGR sendromundan şüpheleniliyorsa genetik test önerilir 2)
QAniridi için genetik test yaptırmalı mıyım?
A
PAX6 gen testi kesin tanıyı doğrulamak için gereklidir ve özellikle sporadik vakalarda Wilms tümörü risk değerlendirmesi için PAX6 ve WT1 genetik testleri önerilir. Testin DNA dizileme ve MLPA/CMA kombinasyonu ile uygun genetik danışmanlık eşliğinde yapılması önemlidir.
Açı displazisi, kalıcı iris kökü ve trabeküler ağ yapışıklıklarının değerlendirilmesi
Göz içi basıncı ölçümü (düzenli)
Glokom taraması. Ergenlikten itibaren düzenli olarak yapılır
Abdominal ultrasonografi
Wilms tümörü taraması (sporadik vakalar, birkaç ayda bir, özellikle 5 yaşına kadar)
Genetik test
PAX6 gen mutasyonu veya 11p13 bölgesi delesyonunun belirlenmesi (kesin tanı için gerekli)
Çocuklarda genel anestezi altında muayene gerekebilir.
QAniridi tanısı nasıl konur?
A
Yarık lamba biyomikroskopisi ile iris displazisi doğrulanır ve OCT ile foveal hipoplazi değerlendirilir. PAX6 gen testi ile kesin tanı mümkündür; sporadik vakalarda WT1 geni de araştırılır. Herpetik iris atrofisi, travmatik iris defekti, iris kolobomu, Rieger anomalisi ve ICE sendromundan ayırt edilmesi önemlidir.
İris displazisi, foveal hipoplazi, mikroftalmi ve nistagmus şu anda müdahale edilemez; temel yaklaşım takiptir. Tedavi edilebilen durumlar keratopati, katarakt, glokom, fotofobi ve az görmedir2).
Korneal stromal opasite: Aniridinin komplikasyonları nedeniyle kornea nakli ile elde edilen görme iyileşmesi sınırlıdır2). Uzun vadede glokomun kötüleşmesi ve greft yetmezliği nedeniyle görme prognozu sıklıkla kötüdür. Kornea opasitesi için tam kat kornea nakli genellikle görme iyileşmesi sağlamaz ve yüksek red reaksiyonu oranına dikkat edilmelidir. Ağır vakalarda, yarar ve zarar dengesi dikkatlice değerlendirildikten sonra uygulamaya karar verilir.
Korneal epitelyal kök hücre yetmezliği (LSCD): Cerrahi tedavi düşünülmelidir2). Spesifik olarak, allojenik limbal transplantasyon (KLAL) veya kültüre oral mukoza epitel transplantasyonu (COMET) ile bir dereceye kadar oküler yüzey rekonstrüksiyonu beklenebilir3). Korneal stromal opasite eşlik ediyorsa, kornea naklinin eklenmesi genellikle görme iyileşmesi için faydalıdır2).
Glokom implant cerrahisi: Uzun tüp cerrahisi (tesis onayı gerekli)
Siliyer cisim fotokoagülasyonu: Diğer tedavilerin başarısız olduğu durumlarda son çare
İlaç tedavisine direnç sık görülür ve tüp şant cerrahisi iyi bir seçenek olabilir4). Glokomda görme alanı hasarı geri dönüşümsüz olduğundan, erken göz içi basıncı kontrolü görme fonksiyonunun korunmasında anahtardır.
QAniridiye bağlı glokom nasıl tedavi edilir?
A
Öncelikle damla ve ağızdan ilaç tedavisi uygulanır, ancak çoğu vaka ilaca dirençlidir. Yetersiz yanıt durumunda dışa akım yollarının yeniden yapılandırılması (goniyotomi, trabekülotomi) düşünülür, ardından trabekülektomi veya uzun tüp cerrahisi (glokom implant cerrahisi) uygulanır. Uzun tüp cerrahisi tesis onayı gerektirir. Siliyer cisim fotokoagülasyonu, diğer tedavilerin başarısız olduğu durumlarda son çaredir. Düzenli göz içi basıncı takibi zorunludur.
PAX6 geni, embriyonik dönemde organ farklılaşmasını yöneten transkripsiyon faktörlerini kodlayan bir ana kontrol genidir. Erken göz tomurcuğundan itibaren eksprese olur ve çeşitli transkripsiyon faktörlerini düzenler. PAX6’nın tek alel kaybı (haplo-yetersizlik), gözün tamamını etkileyen konjenital anomalilere (aniridi, Peters anomalisi, maküler hipoplazi vb.) neden olur.
PAX6 mutasyonları çoğunlukla nonsense, frame-shift gibi PTC tipindedir ve missense mutasyonlar da bildirilmiştir1). Genotip-fenotip korelasyonu üzerine yapılan çalışmalar, mutasyon tipine göre oftalmolojik bulguların şiddetinin farklılık gösterdiğini ortaya koymuştur3).
PAX6, göz dışında merkezi sinir sistemi, pankreas Langerhans adacıkları ve koku epitelinde de eksprese olur; bu dokuların hipoplazisine bağlı olarak göz dışı komplikasyonlar (korpus kallozum agenezisi, epilepsi, anosmi, glukoz intoleransı) ortaya çıkabilir1).
Açık açılı patofizyoloji: Trabeküler ağda aköz hümör çıkış direncinin artması
Kapalı açılı patofizyoloji: En periferde kalan iris kökünün trabeküler ağa yapışması ve bir tür kapalı açılı glokom patofizyolojisi oluşturması
Bebeklik döneminde glokom nadirdir; genellikle büyümeyle birlikte ergenlikte ilerleyici olarak ortaya çıkar. Açı anomalilerine bağlı açık açılı veya kapalı açılı glokom şeklinde olabilir.
Korneal limbal kök hücre yetmezliği (LSCD) patofizyolojisi
Patolojik olarak korneal epitel kök hücre fonksiyon bozukluğu görülür; epitel ve Bowman membranında anormallikler oluşur ve damardan zengin pannus gelişir. Palisades of Vogt’un hipoplazisi, konjonktival doku invazyonu ve keratinizasyona ilerler1).
Aniridili gözlerde kornea sağlıklı bireylere göre daha kalındır. Çocuklukta kornea genellikle normaldir, ancak büyümeyle birlikte korneal stromal opasite ve LSCD gelişerek görme azalmasına neden olur. 14 yıllık tek merkezli bir çalışmada (738 göz), LSCD nedenleri arasında aniridi %30,9 ile en sık görülen nedendi6).
Görme prognozu genellikle kötüdür ve sıklıkla 0,1 düzeyindedir
Maküler hipoplazi için etkili bir tedavi yoktur ve en önemli görme kısıtlayıcı faktördür
Glokoma bağlı görme alanı kaybı geri dönüşümsüzdür; erken göz içi basıncı kontrolü önemlidir
Sporadik vakalarda Wilms tümörünün 5 yaşına kadar erken başlangıcına dikkat edilmeli ve düzenli abdominal ultrasonografiye devam edilmelidir.
Uzun dönem prognozla ilgili çalışmalar, görme prognozunun genellikle kötü olduğunu ancak komplikasyonların türü ve şiddetine göre bireysel farklılıklar olduğunu bildirmektedir5).
Yeni nesil dizilemenin (NGS) yaygınlaşmasıyla, izole aniridide PAX6 mutasyonlarının saptanma oranı yaklaşık %85’e ulaşmıştır2). Kromozomal mikrodizin (CMA), 11p13 mikrodelesyonlarının saptanmasında geleneksel kromozom analizine göre daha duyarlıdır ve WAGR sendromunun tanı doğruluğunu artırmaya katkıda bulunmaktadır2).
Kültüre oral mukoza epitel transplantasyonunun (COMET) uzun dönem sonuçlarına ilişkin veriler birikmektedir2). Boston tip I keratoprotez ile kısa dönemde (17-28,7 ay) %65-93 oranında görme iyileşmesi sağlandığı, ancak 4,5 yılda bu oranın %43,5’e düştüğü bildirilmiştir2).
Yapay İris Cihazları ve Gen Tedavisi Perspektifleri
HumanOptics CustomFlex ArtificialIris, kişiye özel silikon yapay iris cihazıdır ve fotofobi azaltma ile kozmetik iyileştirmede faydalı olduğu düşünülmektedir; ancak 2024 itibarıyla Japonya’da onaylanmamıştır. PAX6 haplo-yetersizliğini hedef alan moleküler hedefli tedaviler şu anda araştırma aşamasındadır ve klinik uygulamaya ulaşmamıştır3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.