先天性并发症(出生时即存在)

无虹膜症(Aniridia)
1. 什么是无虹膜症
Section titled “1. 什么是无虹膜症”无虹膜症(aniridia)是因先天性因素导致虹膜完全或不完全缺失的状态。虽称为“无虹膜”,但房角最周边部常残留虹膜根部。
2017年被日本厚生劳动省根据难病法认定为指定难病1)。被诊断为指定难病且严重程度分级为Ⅲ度及以上的患者可享受医疗费用补助,并根据收入设定个人负担上限2)。
| 项目 | 内容 |
|---|---|
| 患病率 | 每6.4万至9.6万人中有1人1) |
| 性别差异 | 无1) |
| 双眼性 | 60%至90%1) |
| 遗传方式(家族性) | 约占全部病例的2/3(常染色体显性遗传) |
| 散发性 | 约占全部病例的1/3 |
| 合并Wilms肿瘤(散发病例) | 约30%(WAGR综合征)3) |
瑞典和挪威的流行病学研究表明,患病率约为每90,000人中1人3)。对43例携带PAX6基因突变患者的详细眼科评估显示,虹膜发育异常的程度因突变类型而异3)。
Q
无虹膜症会遗传吗?
A
约2/3的病例为常染色体显性遗传,患病父母有50%的概率将疾病遗传给子女。其余1/3为散发性,无家族史。散发性病例有合并Wilms肿瘤(肾肿瘤)的WAGR综合征风险,因此建议进行PAX6基因和WT1基因的遗传学检测。
2. 主要症状与临床所见
Section titled “2. 主要症状与临床所见”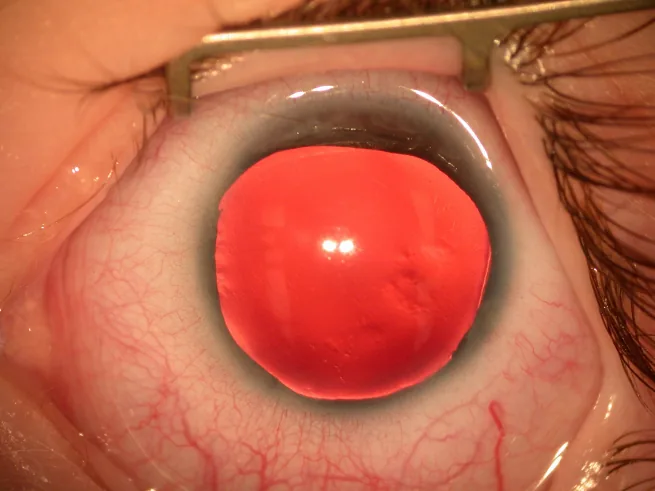
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
由于虹膜缺失或不完整,瞳孔无法正常功能,不能调节进入眼球的光量,因此患者主诉严重畏光。此外,黄斑发育不良导致的固视不良,常成为出生后早期即出现的水平性眼球震颤的主诉。
后天性并发症(随成长发生)
眼并发症频率总结
Section titled “眼并发症频率总结”| 并发症 | 频率·时期 | 对视功能的影响 |
|---|---|---|
| 黄斑发育不良 | 几乎全部(先天性) | 最大视力限制因素。无有效治疗方法 |
| 白内障 | 约80%(后天性)1) | 视力下降、畏光加重 |
| 青光眼 | 50〜75%(后天性)1) | 进展可导致不可逆的视野缺损 |
| 角膜缘干细胞缺乏症 | 成年后发病并进展3) | 角膜实质混浊→严重视力下降 |
| 眼球震颤 | 先天性(几乎全部病例) | 固视不良 |
| 斜视 | 先天性至婴幼儿期 | 弱视风险 |
PAX6基因除眼组织外,还在中枢神经、胰腺朗格汉斯岛、嗅上皮中表达。这些组织的发育不全可能导致多种眼外并发症1)。
- 胼胝体缺失、癫痫、高级脑功能障碍
- 无嗅觉症
- 葡萄糖不耐受
- WAGR综合征(约30%的散发病例):Wilms肿瘤、无虹膜、泌尿生殖器异常、智力障碍3)
Q
无虹膜症患者视力如何?
A
视力预后通常较差,多为0.1左右。但根据黄斑发育不全的程度及有无并发症,个体差异可达0.1至0.7。目前黄斑发育不全尚无有效治疗方法,是视力受限的最大因素。通过适当的屈光矫正和低视力护理,可改善生活质量。
3. 病因与风险因素
Section titled “3. 病因与风险因素”病因:PAX6基因单倍剂量不足
Section titled “病因:PAX6基因单倍剂量不足”无虹膜症的病因是位于第11号染色体短臂(11p13)的PAX6基因单等位基因功能丧失(单倍剂量不足)。功能性基因剂量减半导致发病。双等位基因异常时可能导致胚胎致死1)。
PAX6是胚胎期器官分化的转录因子主控基因,调控多种转录因子。PAX6异常可导致眼球整体的多种先天异常(无虹膜、Peters异常、黄斑发育不全等)。
基因突变类型多为无义突变、移码突变等提前终止密码子(PTC)型突变,也有错义突变的报道1)。孤立性无虹膜症的测序分析中,约85%可检测到PAX6突变2)。
WAGR综合征(散发病例注意事项)
Section titled “WAGR综合征(散发病例注意事项)”PAX6基因与抑癌基因WT1在11p13染色体上相邻。散发病例中,由于相邻基因缺失,可能出现Wilms肿瘤、无虹膜、泌尿生殖器异常、智力障碍(mental retardation)组成的WAGR综合征3)。约30%的散发病例在5岁前早期双侧发生Wilms肿瘤。
遗传咨询要点
Section titled “遗传咨询要点”- PAX6突变阳性且无WT1缺失→可推测无WAGR综合征的可能性2)
- 遗传学检查应结合DNA测序与MLPA/CMA检测基因组结构异常2)
- 对于疑似WAGR综合征的散发病例,建议进行遗传学检测2)
Q
是否应该接受无虹膜症的基因检测?
A
PAX6基因检测对于确诊Definite诊断是必要的,尤其是散发病例,为评估Wilms肿瘤风险,建议进行PAX6和WT1的遗传学检测。检测应结合DNA测序和MLPA/CMA,并在适当的遗传咨询下进行。
4. 诊断与检查方法
Section titled “4. 诊断与检查方法”诊断标准(厚劳省指定难病 2020)
Section titled “诊断标准(厚劳省指定难病 2020)”无虹膜症的诊断标准及严重程度分类1)的类别分类如下所示。
| 诊断类别 | 诊断标准的组合 |
|---|---|
| Definite | 满足A中任意一项+B1+E,并排除C |
| Probable (1) | 满足A中任意一项+B1+F,并排除C |
| Probable (2) | 满足A中任意一项+B1+B2,并排除C |
| Probable (3) | 满足A中任意一项+B1+B3,并排除C |
| 可能 | 满足A中任意一项+B1,且不能完全排除C |
A. 症状
B. 检查所见
- 裂隙灯显微镜检查显示虹膜发育异常,从部分虹膜萎缩到完全虹膜缺损不等(60-90%为双眼)
- 眼底检查/OCT检查显示黄斑发育不良(中心凹凹陷、黄斑色素、中心凹无血管区不明显)
- 裂隙灯显微镜检查显示角膜缘干细胞缺乏、角膜混浊等角膜病变
- 裂隙灯显微镜检查显示白内障(约80%合并)
- 超声检查/MRI/CT显示小眼球
- 眼球震颤
- 眼压检查等显示青光眼(50-75%合并)
C. 鉴别诊断(应排除的疾病)
D. PAX6基因突变相关的眼外并发症(胼胝体缺损、癫痫等)
E. PAX6基因的致病性基因突变或11p13区域缺失(遗传学检查)
F. 家族内发病(常染色体显性遗传占2/3)
标准检查方法
Section titled “标准检查方法”| 检查 | 目的·内容 |
|---|---|
| 裂隙灯显微镜检查 | 虹膜发育异常程度评估(诊断基础) |
| 眼底检查·OCT | 黄斑发育不良(中心凹凹陷消失·黄斑色素不明显)的评估 |
| 房角镜检查 | 房角发育不全·残留虹膜根部与小梁网粘连的评估 |
| 眼压测量(定期) | 青光眼筛查。从青春期开始定期进行 |
| 腹部超声检查 | Wilms肿瘤筛查(散发病例,每数月一次,尤其5岁前) |
| 基因检测 | 鉴定PAX6基因突变或11p13区域缺失(确诊所需) |
儿童可能需要在全身麻醉下进行检查。
Q
无虹膜症如何诊断?
5. 标准治疗方法
Section titled “5. 标准治疗方法”治疗总体方针
Section titled “治疗总体方针”虹膜发育异常、黄斑发育不良、小眼球、眼球震颤目前无法干预,基本以观察为主。治疗对象为角膜病变、白内障、青光眼、畏光、低视力2)。
治疗方针概要
Section titled “治疗方针概要”| 治疗领域 | 诊疗方针 |
|---|---|
| 角膜实质混浊 | 角膜移植对视功能改善有限,需谨慎判断适应症 |
| 角膜上皮干细胞衰竭症 | 考虑眼表面重建术 |
| 白内障 | 根据混浊和畏光程度考虑手术 |
| 高眼压·青光眼 | 为保存视功能积极治疗 |
| 低视力护理 | 早期引入 |
| 畏光 | 使用遮光眼镜或隐形眼镜等措施 |
角膜病变的治疗
Section titled “角膜病变的治疗”角膜实质混浊:由于无虹膜症的并发症,通过角膜移植获得的视功能改善有限2)。长期来看,因青光眼恶化及移植片随时间功能不全,视力预后往往不良。全层角膜移植治疗角膜混浊常不能改善视力,且需注意排斥反应率高。重症病例应在充分权衡利弊后决定是否施行。
角膜上皮干细胞缺乏症(LSCD):考虑手术治疗2)。具体而言,通过异体角膜缘移植(KLAL)或培养口腔黏膜上皮移植(COMET)可在一定程度上重建眼表3)。若合并角膜实质混浊,联合角膜移植常有助于提高视力2)。
白内障的治疗
Section titled “白内障的治疗”20岁前50%~85%的患者发生白内障,根据混浊和畏光程度计划白内障手术2)。
- 晶状体囊及Zinn小带脆弱导致手术难度高
- 注意术后青光眼恶化、前部纤维化综合征及大疱性角膜病变的风险2)
- 人工晶状体(IOL)植入需谨慎适应3)
- 不建议在白内障手术同时植入人工虹膜,因其可能诱发青光眼
在充分说明手术相关风险后实施。
青光眼的治疗
Section titled “青光眼的治疗”青光眼直接影响视功能预后,应积极治疗2)。采取以下分步方法。
- 药物治疗:注意副作用,考虑对儿童的全身影响,通过滴眼液和口服药物降低眼压
- 流出道重建术:房角切开术、小梁切开术(药物治疗无效时考虑)
- 滤过手术:小梁切除术
- 青光眼植入手术:长管手术(需机构认证)
- 睫状体凝固术:其他治疗无效时的最后手段
药物治疗常耐药,管分流手术可能是较好的选择4)。青光眼导致的视野损害不可逆,因此早期眼压管理是维持视功能的关键。
Q
无虹膜症的青光眼如何治疗?
A
首先进行滴眼液和口服药物疗法,但多数患者对药物耐药。效果不佳时考虑流出道重建术(房角切开术、小梁切开术),进一步可进行小梁切除术或长管手术(青光眼植入手术)。长管手术需机构认证。睫状体凝固术是其他治疗无效时的最后手段。定期监测眼压至关重要。
低视力护理与畏光对策
Section titled “低视力护理与畏光对策”低视力护理和畏光对策应尽早引入,以维持视功能和生活质量2)。
- 屈光矫正:用眼镜矫正屈光不正,尽可能促进视觉发育(基础)
- 遮光眼镜:有效减轻畏光。严重畏光时处方
- 带人工虹膜的SCL:改善畏光和外观
- 使用放大镜、弱视眼镜、放大阅读器等视觉辅助器具
6. 病理生理学·详细发病机制
Section titled “6. 病理生理学·详细发病机制”PAX6基因与眼形成
Section titled “PAX6基因与眼形成”PAX6基因是编码胚胎期器官分化转录因子的主控基因。它在早期眼球中表达,调控多种转录因子。PAX6单等位基因功能丧失(单倍剂量不足)会导致全眼球先天性异常(如无虹膜、Peters异常、黄斑发育不良等)。
PAX6突变多为无义或移码等PTC型,也有错义突变的报道1)。关于基因型-表型相关性的研究表明,突变类型不同,眼科表现的严重程度也不同3)。
PAX6除眼球外,还在中枢神经、胰腺朗格汉斯岛、嗅上皮中表达,各组织发育不良可能导致眼外并发症(如胼胝体缺失、癫痫、嗅觉缺失、葡萄糖不耐受)1)。
青光眼的发病机制
Section titled “青光眼的发病机制”无虹膜症伴发青光眼的发病机制有两种途径。
- 开角型病变:小梁网房水流出阻力增加
- 闭角型病变:最周边残留的虹膜根部与小梁网粘连,导致一种闭角型青光眼病变
婴儿期出现青光眼罕见,随成长在青年期进行性发病。可能因房角发育异常呈开放状态发病,或因房角关闭导致青光眼。
角膜缘干细胞缺乏症(LSCD)的病理
Section titled “角膜缘干细胞缺乏症(LSCD)的病理”病理学上可见角膜上皮干细胞功能异常,导致上皮和Bowman膜异常,形成富含血管的角膜血管翳。从Vogt栅栏发育不全进展为结膜组织侵入和角化1)。
无虹膜症的角膜比健康人厚。幼年时角膜多正常,随成长合并角膜实质混浊和LSCD,导致视力下降。一项14年单中心研究(738眼)显示,在LSCD的病因中,无虹膜症占30.9%,为最多6)。
- 视力预后通常不良,多为0.1左右
- 黄斑发育不良无有效治疗方法,是最大的视力限制因素
- 青光眼导致的视野损害不可逆,早期眼压管理至关重要
- 对于散发病例,需注意Wilms肿瘤在5岁前的早期发病,并定期进行腹部超声检查
关于长期预后的研究报告显示,视力预后总体不佳,但根据并发症的类型和严重程度存在个体差异5)。
7. 最新研究与未来展望
Section titled “7. 最新研究与未来展望”遗传学检查的进展
Section titled “遗传学检查的进展”随着下一代测序(NGS)的普及,孤立性无虹膜症中PAX6突变的检出率约为85%2)。染色体微阵列分析(CMA)在检测11p13微缺失方面比传统染色体检查更敏感,有助于提高WAGR综合征的诊断准确性2)。
培养口腔黏膜上皮移植(COMET)的长期结果数据正在积累中2)。关于Boston I型人工角膜,短期(17-28.7个月)内65-93%的患者视力改善,但4.5年时降至43.5%2)。
人工虹膜装置与基因治疗展望
Section titled “人工虹膜装置与基因治疗展望”HumanOptics CustomFlex ArtificialIris是一种定制硅胶人工虹膜装置,有助于减轻畏光和改善外观,但截至2024年在日本尚未获批。针对PAX6单倍剂量不足的分子靶向治疗目前处于研究阶段,尚未进入临床应用3)。
8. 参考文献
Section titled “8. 参考文献”- 大家義則, 川崎諭, 西田希, 木下茂, 外園千恵, 大橋裕一, 他. 無虹彩症の診断基準および重症度分類. 日眼会誌. 2020;124:83-88.
- 厚生労働科学研究費補助金難治性疾患政策研究事業「角膜難病の標準的診断法および治療法の確立を目指した調査研究」研究班. 無虹彩症の診療ガイドライン. 日眼会誌. 2021;125:38-73.
- Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
- American Academy of Ophthalmology. Diagnosis and Management of Aniridia. EyeNet Magazine. 2014. https://www.aao.org/eyenet/article/diagnosis-management-of-aniridia
- Japanese Ophthalmological Society. Clinical practice guideline for aniridia. Jpn J Ophthalmol. 2026. doi:10.1007/s10384-025-01296-y. https://link.springer.com/article/10.1007/s10384-025-01296-y
- Hu JCW, Weissbart SB. Limbal stem cell deficiency and severe ocular surface disease: a review. Ann Eye Sci. 2023;8:35.