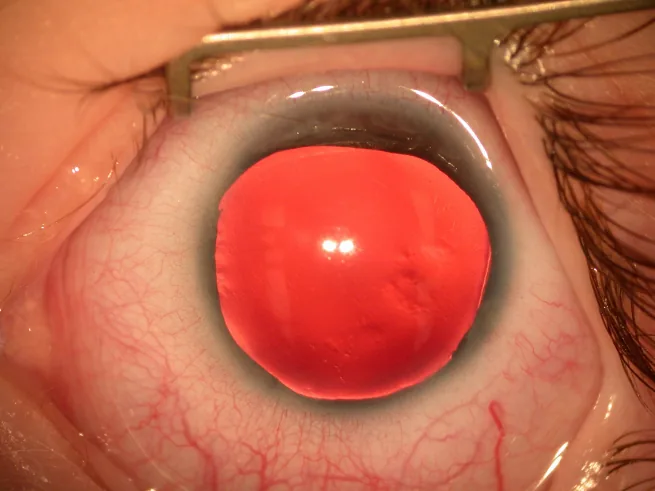
La aniridia es una condición en la que el iris está total o parcialmente ausente debido a factores congénitos. Aunque se denomina “aniridia”, a menudo queda un remanente de la raíz del iris en la periferia del ángulo.
En 2017 fue designada como enfermedad rara según la Ley de Enfermedades Raras del Ministerio de Salud, Trabajo y Bienestar de Japón 1). Los pacientes diagnosticados con esta enfermedad designada y clasificados con gravedad de grado III o superior son elegibles para asistencia financiera médica, con un límite máximo de copago según los ingresos 2).
Aproximadamente 2/3 del total (autosómico dominante)
Esporádico
Aproximadamente 1/3 del total
Asociación con tumor de Wilms (casos esporádicos)
Aproximadamente 30% (síndrome WAGR)3)
Los estudios epidemiológicos en Suecia y Noruega reportan una prevalencia de aproximadamente 1 en 90,000 personas3). Una evaluación oftalmológica detallada de 43 casos con mutaciones en el gen PAX6 mostró que el grado de anomalía del iris varía según el tipo de mutación3).
Q¿La aniridia es hereditaria?
A
Alrededor de dos tercios de los casos son hereditarios autosómicos dominantes, con un 50% de probabilidad de transmitirse de un padre afectado a un hijo. El tercio restante son casos esporádicos sin antecedentes familiares. En los casos esporádicos, existe riesgo de síndrome WAGR, que incluye tumor de Wilms (tumor renal), por lo que se recomiendan pruebas genéticas de los genes PAX6 y WT1.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Fotografía con lámpara de hendidura del segmento anterior que muestra el iris casi ausente, con solo un borde muy delgado de iris residual visible en la periferia. Muestra directamente los hallazgos clínicos típicos de la aniridia, adecuada para explicar los principales síntomas y hallazgos clínicos.
Debido a que el iris está ausente o incompleto, la pupila no funciona y no puede regular la cantidad de luz que entra al ojo. Por lo tanto, los pacientes se quejan de fotofobia intensa. Además, la mala fijación debido a la hipoplasia macular a menudo se presenta como queja principal de nistagmo horizontal desde la primera infancia.
Fotofobia: Incapacidad para regular la luz debido a la ausencia del iris → deslumbramiento intenso
Nistagmo (nistagmo horizontal): Mala fijación por hipoplasia macular. Aparece desde la primera infancia
Baja visión: Factores combinados de hipoplasia macular, catarata, glaucoma y deficiencia límbica corneal (LSCD)
Anomalía del iris: Desde atrofia parcial hasta ausencia completa, con diversos grados
Hipoplasia macular: Presente en casi todos los casos. Ausencia de la fóvea y pigmentación macular poco clara. Es el factor limitante más importante para la visión
Complicaciones adquiridas (aparecen con el crecimiento)
Cataratas: presentes en aproximadamente el 80%. Se desarrollan en un 50-85% antes de los 20 años
Glaucoma: presente en un 50-75%. Es raro en la infancia y progresa en la adolescencia
Deficiencia de células madre limbares (LSCD): suele ser normal en la infancia, pero con el crecimiento progresa la opacidad del estroma corneal y el pannus vascular
Resumen de la frecuencia de las complicaciones oculares
El gen PAX6 se expresa no solo en tejidos oculares, sino también en el sistema nervioso central, los islotes de Langerhans del páncreas y el epitelio olfatorio. La hipoplasia de estos tejidos puede provocar diversas complicaciones extraoculares1).
Agenesia del cuerpo calloso, epilepsia, disfunción cognitiva superior
Anosmia
Intolerancia a la glucosa
Síndrome WAGR (aproximadamente 30% de los casos esporádicos): tumor de Wilms, aniridia, anomalías genitourinarias, retraso mental3)
Q¿Cuánto se puede ver con aniridia?
A
El pronóstico visual generalmente es malo, con una agudeza visual de alrededor de 0.1. Sin embargo, puede variar de 0.1 a 0.7 según el grado de hipoplasia macular y la presencia de complicaciones. Actualmente no existe un tratamiento eficaz para la hipoplasia macular, que es el factor limitante más importante de la visión. La calidad de vida diaria puede mejorar con una corrección refractiva adecuada y cuidados de baja visión.
La aniridia es causada por la pérdida de función de un alelo (haploinsuficiencia) del gen PAX6, ubicado en el brazo corto del cromosoma 11 (11p13). Ocurre debido a la reducción a la mitad de la dosis génica funcional. Se cree que la anomalía de ambos alelos es letal en el período embrionario1).
PAX6 es un gen maestro de control que codifica un factor de transcripción responsable de la diferenciación de órganos en el período embrionario, y regula diversos factores de transcripción. Las anomalías de PAX6 causan diversas malformaciones congénitas en todo el globo ocular (aniridia, anomalía de Peters, hipoplasia macular, etc.).
Los tipos de mutaciones genéticas son con frecuencia mutaciones de tipo codón truncado prematuro (PTC), como sin sentido y cambio de marco de lectura, aunque también se han reportado mutaciones de sentido erróneo1). En el análisis de secuenciación de aniridia aislada, se detectan mutaciones de PAX6 en aproximadamente el 85% de los casos2).
Síndrome WAGR (consideraciones en casos esporádicos)
El gen PAX6 se encuentra adyacente al gen supresor de tumores WT1 en el cromosoma 11p13. En casos esporádicos, la deleción de genes contiguos puede dar lugar al síndrome WAGR, que comprende tumor de Wilms, aniridia, anomalías genitourinarias y retraso mental3). Aproximadamente el 30% de los casos esporádicos desarrollan tumor de Wilms bilateral temprano antes de los 5 años.
Mutación de PAX6 positiva sin deleción de WT1 → se puede inferir que no hay probabilidad de síndrome WAGR2)
Las pruebas genéticas combinan secuenciación de ADN con detección de anomalías estructurales del genoma mediante MLPA/CMA2)
En casos esporádicos con sospecha de síndrome WAGR, se recomienda realizar pruebas genéticas 2)
Q¿Debo realizarme pruebas genéticas para la aniridia?
A
La prueba genética de PAX6 es necesaria para confirmar el diagnóstico definitivo, y en casos esporádicos se recomienda el análisis genético de PAX6 y WT1 para evaluar el riesgo de tumor de Wilms. Es importante realizar la prueba combinando secuenciación de ADN y MLPA/CMA, bajo asesoramiento genético adecuado.
Detección de glaucoma. Realizar periódicamente desde la adolescencia
Ecografía abdominal
Detección de tumor de Wilms (casos esporádicos, cada pocos meses, especialmente hasta los 5 años)
Prueba genética
Identificación de mutaciones en PAX6 o deleción de la región 11p13 (necesaria para el diagnóstico definitivo)
En niños, puede ser necesario realizar pruebas bajo anestesia general.
Q¿Cómo se realiza el diagnóstico de aniridia?
A
El diagnóstico básico consiste en confirmar la anomalía del iris mediante examen con lámpara de hendidura y evaluar la hipoplasia foveal con OCT. El diagnóstico definitivo es posible mediante la prueba genética de PAX6, y en casos esporádicos también se realiza la búsqueda del gen WT1. Es importante diferenciar de atrofia del iris por herpes, defecto del iris postraumático, coloboma del iris, anomalía de Rieger y síndrome ICE.
La anomalía del iris, la hipoplasia foveal, la microftalmía y el nistagmo no son intervenibles en la actualidad, por lo que la observación es la base. Los objetivos del tratamiento son la queratopatía, la catarata, el glaucoma, la fotofobia y la baja visión2).
Opacidad del estroma corneal: La mejora de la función visual obtenida mediante trasplante de córnea es limitada debido a las complicaciones de la aniridia2). A largo plazo, el pronóstico visual suele ser desfavorable debido al empeoramiento del glaucoma y al fracaso del injerto con el tiempo. El trasplante de córnea de espesor total para la opacidad corneal a menudo no conduce a una mejora visual y se debe tener en cuenta la alta tasa de rechazo. En casos graves, se debe evaluar la relación beneficio-riesgo antes de realizarlo.
Insuficiencia de células madre del epitelio corneal (LSCD): Se debe considerar el tratamiento quirúrgico2). Específicamente, el trasplante de limbo alogénico (KLAL) o el trasplante de epitelio oral cultivado (COMET) pueden ofrecer cierta reconstrucción de la superficie ocular3). Cuando se combina con opacidad del estroma corneal, el trasplante de córnea concomitante suele ser útil para mejorar la agudeza visual2).
Entre el 50 y el 85% de los pacientes desarrollan cataratas antes de los 20 años, y la cirugía de cataratas se planifica según la intensidad de la opacidad y la fotofobia2).
Alta dificultad quirúrgica debido a la fragilidad del saco capsular y las zónulas de Zinn
Tener en cuenta el riesgo de empeoramiento del glaucoma postoperatorio, síndrome de fibrosis anterior y queratopatía bullosa2)
La inserción de lente intraocular (LIO) requiere una indicación cuidadosa3)
No se recomienda la inserción simultánea de un iris artificial durante la cirugía de cataratas, ya que puede inducir glaucoma
Realizar después de informar adecuadamente sobre los riesgos asociados a la cirugía.
El glaucoma afecta directamente el pronóstico visual, por lo que se debe tratar activamente2). Se sigue un enfoque escalonado:
Tratamiento farmacológico: Reducción de la presión intraocular mediante gotas o medicación oral, teniendo en cuenta los efectos secundarios y el impacto sistémico en niños
Cirugía de reconstrucción de la vía de drenaje: Goniotomía o trabeculotomía (considerar si el tratamiento farmacológico es ineficaz)
Cirugía de implante de glaucoma: cirugía de tubo largo (requiere acreditación del centro)
Cicloablación: último recurso cuando otros tratamientos no funcionan
La resistencia al tratamiento farmacológico es frecuente, y la cirugía de derivación con tubo puede ser una buena opción4). Dado que el daño del campo visual en el glaucoma es irreversible, el control temprano de la presión intraocular es clave para preservar la función visual.
Q¿Cómo se trata el glaucoma en la aniridia?
A
Primero se realiza tratamiento farmacológico con gotas y medicación oral, pero a menudo es resistente. Si es insuficiente, se considera la cirugía reconstructiva de la vía de drenaje (goniotomía/trabeculotomía), y luego se avanza a trabeculectomía o cirugía de tubo largo (implante de glaucoma). La cirugía de tubo largo requiere acreditación del centro. La cicloablación es el último recurso cuando otros tratamientos no funcionan. Es esencial un monitoreo regular de la presión intraocular.
El gen PAX6 es un gen maestro de control que codifica un factor de transcripción que regula la diferenciación de órganos durante el período embrionario. Se expresa desde el ojo primitivo y coordina varios factores de transcripción. La pérdida de función de un alelo de PAX6 (haploinsuficiencia) causa anomalías congénitas en todo el ojo (aniridia, anomalía de Peters, hipoplasia macular, etc.).
Las mutaciones de PAX6 son a menudo de tipo PTC, como sin sentido y cambio de marco, y también se han reportado mutaciones de sentido erróneo 1). Los estudios sobre la correlación genotipo-fenotipo muestran que la gravedad de los hallazgos oftalmológicos varía según el tipo de mutación 3).
PAX6 también se expresa fuera del ojo, en el sistema nervioso central, los islotes de Langerhans del páncreas y el epitelio olfatorio, y puede causar complicaciones extraoculares (agenesia del cuerpo calloso, epilepsia, anosmia, intolerancia a la glucosa) debido a la hipoplasia de estos tejidos 1).
Se consideran dos vías en el mecanismo de desarrollo del glaucoma asociado a la aniridia.
Patología de ángulo abierto: aumento de la resistencia al flujo de humor acuoso en la malla trabecular
Patología de ángulo cerrado: la raíz del iris residual en la parte más periférica se adhiere a la malla trabecular, causando una especie de glaucoma de ángulo cerrado
Es raro que se presente glaucoma en la infancia; generalmente se desarrolla de forma progresiva en la adolescencia con el crecimiento. Puede ocurrir en estado abierto debido a una malformación del ángulo, o presentarse como glaucoma de ángulo cerrado.
Patología de la deficiencia de células madre del limbo corneal (LSCD)
Patológicamente, se observa una disfunción de las células madre del epitelio corneal, con anomalías en el epitelio y la membrana de Bowman, y formación de un pannus vascularizado. La hipoplasia de las palizadas de Vogt progresa hacia la invasión y queratinización del tejido conjuntival 1).
La córnea en la aniridia es más gruesa que en individuos sanos. A menudo la córnea es normal en la infancia, pero con el crecimiento se desarrollan opacidad del estroma corneal y LSCD, causando disminución de la agudeza visual. En un estudio unicéntrico de 14 años (738 ojos), la aniridia fue la causa más frecuente de LSCD, representando el 30.9% 6).
El pronóstico visual es generalmente desfavorable, a menudo alrededor de 0.1
La hipoplasia macular no tiene tratamiento efectivo y es el factor limitante más importante de la agudeza visual
El daño del campo visual por glaucoma es irreversible, por lo que el control temprano de la presión intraocular es importante
En casos esporádicos, preste atención a la aparición temprana del tumor de Wilms antes de los 5 años y continúe con ecografías abdominales periódicas.
Los estudios sobre el pronóstico a largo plazo informan que el pronóstico visual es generalmente desfavorable, pero varía según el tipo y la gravedad de las complicaciones5).
Con la difusión de la secuenciación de nueva generación (NGS), la tasa de detección de mutaciones de PAX6 en aniridia aislada es de aproximadamente el 85%2). Los microarrays cromosómicos (CMA) son más sensibles que las pruebas cromosómicas convencionales para detectar microdeleciones en 11p13, lo que contribuye a mejorar la precisión diagnóstica del síndrome de WAGR2).
Se están acumulando resultados a largo plazo del trasplante de mucosa oral cultivada (COMET)2). En cuanto a la queratoprótesis tipo Boston I, se ha informado que la agudeza visual mejora en un 65-93% a corto plazo (17-28.7 meses), pero disminuye al 43.5% a los 4.5 años2).
Perspectivas de los dispositivos de iris artificial y la terapia génica
El HumanOptics CustomFlex ArtificialIris es un dispositivo de iris artificial de silicona hecho a medida, útil para reducir la fotofobia y mejorar la apariencia, pero no está aprobado en Japón a partir de 2024. La terapia dirigida molecularmente contra la haploinsuficiencia de PAX6 se encuentra actualmente en fase de investigación y aún no ha llegado a la aplicación clínica3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.