ภาวะไม่มีม่านตา (Aniridia) เป็นภาวะที่ม่านตา ขาดหายไปทั้งหมดหรือบางส่วนตั้งแต่กำเนิด โดยมีความชุกประมาณ 1 ใน 64,000–96,000 คน และจัดเป็นโรคหายากที่ได้รับการกำหนด
ประมาณ 2 ใน 3 ของผู้ป่วยทั้งหมดเกิดจากการถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่น (ยีน PAX6) ส่วนผู้ป่วยที่เกิดขึ้นเองประมาณ 30% มีความเสี่ยงต่อกลุ่มอาการ WAGR ซึ่งอาจร่วมกับเนื้องอกวิลมส์
ภาวะจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ (Foveal hypoplasia) พบได้เกือบทุกกรณี และเป็นปัจจัยจำกัดการมองเห็น ที่สำคัญที่สุด ปัจจุบันยังไม่มีวิธีการรักษาที่ได้ผล
มักเกิดร่วมกับต้อกระจก (ประมาณ 80%) ต้อหิน (50–75%) และภาวะพร่องเซลล์ขอบกระจกตา (Limbal stem cell deficiency) ในอัตราสูง ทำให้เกิดความบกพร่องทางการมองเห็น ที่ซับซ้อน
ตามการจำแนกความรุนแรง (ระดับ I–IV) ผู้ป่วยที่มีระดับ III ขึ้นไปจะมีสิทธิ์ได้รับความช่วยเหลือด้านค่ารักษาพยาบาล
การรักษาโรคต้อหิน ใช้แนวทางแบบเป็นขั้นตอน โดยเริ่มจากการใช้ยา และในกรณีที่ไม่ได้ผลจะดำเนินการจนถึงการผ่าตัดใส่ท่อระบายน้ำแบบยาว
การดูแลผู้มีสายตาเลือนราง (การแก้ไขสายตา แว่นกันแสง คอนแทคเลนส์ชนิดพิเศษที่มีม่านตาเทียม เป็นต้น) มีความจำเป็นต่อการรักษาการมองเห็น และคุณภาพชีวิต ภาวะไม่มีม่านตา (aniridia) คือภาวะที่ม่านตา ขาดหายไปทั้งหมดหรือบางส่วนเนื่องจากปัจจัยทางพันธุกรรมแต่กำเนิด แม้จะเรียกว่า “ไม่มีม่านตา ” แต่โดยส่วนใหญ่มักมีรากม่านตา หลงเหลืออยู่บริเวณขอบสุดของมุมตา
ในปี พ.ศ. 2560 กระทรวงสาธารณสุข แรงงาน และสวัสดิการของญี่ปุ่นได้กำหนดให้เป็นโรคหายากที่ต้องได้รับการดูแลเป็นพิเศษตามกฎหมายโรคหายาก1) ผู้ป่วยที่ได้รับการวินิจฉัยว่าเป็นโรคหายากและมีระดับความรุนแรงตั้งแต่ระดับ III ขึ้นไปจะมีสิทธิ์ได้รับการช่วยเหลือค่ารักษาพยาบาล โดยกำหนดวงเงินสูงสุดที่ต้องจ่ายเองตามรายได้2)
หัวข้อ เนื้อหา ความชุก 1 ใน 64,000 ถึง 96,000 คน1) ความแตกต่างทางเพศ ไม่มี1) เป็นทั้งสองตา 60–90%1) รูปแบบการถ่ายทอดทางพันธุกรรม (แบบครอบครัว) ประมาณ 2 ใน 3 ของทั้งหมด (ถ่ายทอดแบบ autosomal dominant) ประปราย ประมาณ 1 ใน 3 ของทั้งหมด ร่วมกับเนื้องอก Wilms (กรณีประปราย) ประมาณ 30% (กลุ่มอาการ WAGR) 3)
การศึกษาในสวีเดนและนอร์เวย์รายงานความชุกประมาณ 1 ใน 90,000 คน 3) การประเมินทางจักษุวิทยาอย่างละเอียดในผู้ป่วย 43 รายที่มีการกลายพันธุ์ของยีน PAX6 แสดงให้เห็นว่าระดับความผิดปกติของม่านตา ขึ้นอยู่กับชนิดของการกลายพันธุ์ 3)
Q
โรคแอนิริเดียถ่ายทอดทางพันธุกรรมหรือไม่?
A
ประมาณ 2 ใน 3 ของผู้ป่วยทั้งหมดเป็นแบบถ่ายทอดทางพันธุกรรมแบบออโตโซมัลเด่น ซึ่งมีความเป็นไปได้ 50% ที่จะถ่ายทอดจากพ่อแม่ที่ป่วยไปยังบุตร ส่วนที่เหลืออีก 1 ใน 3 เป็นแบบประปรายโดยไม่มีประวัติครอบครัว ในกรณีที่เป็นแบบประปรายมีความเสี่ยงต่อโรค WAGR syndrome ซึ่งมีเนื้องอกวิลมส์ (เนื้องอกไต) ร่วมด้วย ดังนั้นจึงแนะนำให้ตรวจทางพันธุกรรมของยีน PAX6 และ WT1
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
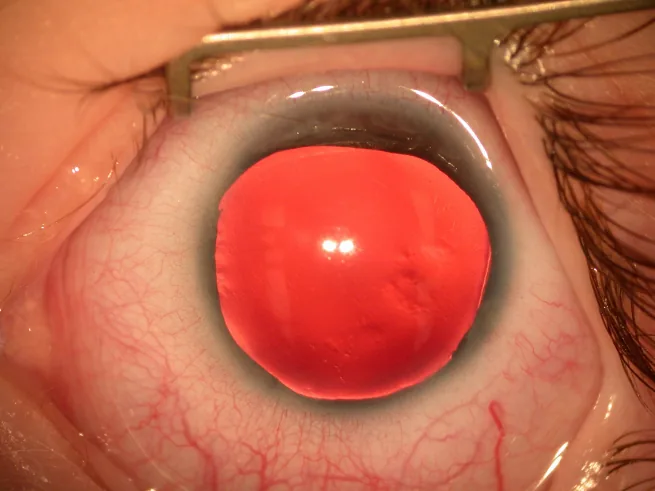
ภาพถ่ายจากกล้องจุลทรรศน์ชนิดกรีดของส่วนหน้าของตา แสดงให้เห็นม่านตา เกือบหายไป เห็นเพียงขอบม่านตา ที่บางมากบริเวณรอบนอก ภาพนี้แสดงอาการแสดงทางคลินิกที่สำคัญของโรคแอนิริเดียโดยตรง เหมาะสำหรับอธิบายอาการหลักและอาการแสดงทางคลินิก
เนื่องจากม่านตา ขาดหรือไม่สมบูรณ์ รูม่านตา จึงไม่ทำงาน ไม่สามารถควบคุมปริมาณแสงที่เข้าสู่ลูกตาได้ ทำให้ผู้ป่วยมีอาการกลัวแสง อย่างรุนแรง นอกจากนี้ การมองเห็น ที่ไม่คงที่เนื่องจากจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ มักเป็นสาเหตุหลักของอาการตากระตุก ในแนวราบที่พบตั้งแต่ช่วงแรกของชีวิต
กลัวแสง : ม่านตา ไม่สามารถควบคุมปริมาณแสงได้ → แสบตาอย่างรุนแรงตากระตุก (แนวราบ)การมองเห็น ไม่คงที่เนื่องจากจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ เกิดขึ้นตั้งแต่ช่วงแรกของชีวิตสายตาเลือนราง : เกิดจากหลายปัจจัยร่วมกัน ได้แก่ จอประสาทตา ส่วนกลางเจริญไม่เต็มที่ ต้อกระจก ต้อหิน และภาวะขอบกระจกตา เสื่อม (LSCD )
ภาวะแทรกซ้อนแต่กำเนิด (ตั้งแต่แรกเกิด)
ความผิดปกติของม่านตา : มีระดับตั้งแต่ฝ่อบางส่วนจนถึงขาดหายทั้งหมด
ภาวะจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ : พบได้เกือบทุกกรณี รอยบุ๋มจอประสาทตา หายไป เม็ดสีจอประสาทตา ส่วนกลางไม่ชัดเจน เป็นปัจจัยจำกัดการมองเห็น ที่สำคัญที่สุด
อาตา อาตา ในแนวราบ เกิดจากภาวะจอประสาทตา ส่วนกลางเจริญไม่เต็มที่
ตาเหล่
ภาวะแทรกซ้อนที่เกิดขึ้นภายหลัง (เกิดขึ้นเมื่อโตขึ้น)
ต้อกระจก
ต้อหิน
ภาวะพร่องเซลล์ต้นกำเนิดลิมบัส (LSCD ) : มักปกติในวัยเด็ก แต่เมื่อโตขึ้นจะเกิดความขุ่นของเนื้อกระจกตา และหลอดเลือดงอกผิดปกติ
ภาวะแทรกซ้อน ความถี่/ระยะเวลา ผลต่อการมองเห็น ภาวะจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ เกือบทั้งหมด (แต่กำเนิด) ปัจจัยจำกัดการมองเห็น สูงสุด ไม่มีการรักษาที่ได้ผล ต้อกระจก ประมาณ 80% (ที่ได้มา) 1) การมองเห็น ลดลง อาการกลัวแสง แย่ลงต้อหิน 50–75% (ที่ได้มา) 1) หากดำเนินไปจะเกิดความบกพร่องของลานสายตา ที่ไม่สามารถกลับคืนได้ ภาวะพร่องเซลล์ต้นกำเนิดลิมบัส เกิดและดำเนินโรคหลังเจริญเติบโต3) ความขุ่นของเนื้อกระจกตา → การมองเห็น ลดลงอย่างรุนแรง อาตา แต่กำเนิด (เกือบทั้งหมด) การจ้องมองไม่ดี ตาเหล่ ตั้งแต่กำเนิดถึงวัยทารก ความเสี่ยงต่อภาวะตาขี้เกียจ
ยีน PAX6 แสดงออกในเนื้อเยื่อนอกเหนือจากตา เช่น ระบบประสาทส่วนกลาง เซลล์ไอส์เลตของลังเกอร์ฮานส์ในตับอ่อน และเยื่อบุรับกลิ่น การเจริญไม่เต็มที่ของเนื้อเยื่อเหล่านี้อาจทำให้เกิดภาวะแทรกซ้อนนอกตาที่หลากหลาย1)
การขาดคอร์ปัสคัลโลซัม โรคลมชัก ความบกพร่องทางการทำงานของสมองระดับสูง
ภาวะเสียการรับกลิ่น
ภาวะไม่ทนต่อกลูโคส
กลุ่มอาการ WAGR (ประมาณ 30% ของผู้ป่วยประปราย): เนื้องอกวิลมส์ ภาวะไม่มีม่านตา ความผิดปกติของอวัยวะสืบพันธุ์และทางเดินปัสสาวะ ภาวะบกพร่องทางสติปัญญา3)
Q
ผู้ป่วยภาวะไม่มีม่านตามองเห็นได้มากน้อยเพียงใด?
A
การพยากรณ์โรคทางสายตาโดยทั่วไปไม่ดี มักอยู่ที่ประมาณ 0.1 อย่างไรก็ตาม ขึ้นอยู่กับระดับของภาวะจอประสาทตา ดำบกพร่องและการมีภาวะแทรกซ้อน อาจมีความแตกต่างระหว่างบุคคลตั้งแต่ 0.1 ถึง 0.7 ปัจจุบันยังไม่มีวิธีการรักษาที่ได้ผลสำหรับภาวะจอประสาทตา ดำบกพร่อง ซึ่งเป็นปัจจัยจำกัดการมองเห็น สูงสุด การแก้ไขสายตาอย่างเหมาะสมและการดูแลผู้มีความบกพร่องทางการมองเห็น สามารถช่วยปรับปรุงคุณภาพชีวิตประจำวันได้
สาเหตุของภาวะไม่มีม่านตา เกิดจากการสูญเสียการทำงานของอัลลีลหนึ่งของยีน PAX6 ซึ่งอยู่บนแขนสั้นของโครโมโซมคู่ที่ 11 (11p13) หรือที่เรียกว่า haploinsufficiency เกิดจากปริมาณยีนที่ทำงานได้ลดลงครึ่งหนึ่ง หากอัลลีลทั้งสองผิดปกติ เชื่อว่าทำให้ทารกในครรภ์เสียชีวิตได้ 1)
PAX6 เป็นยีนควบคุมหลัก (master control gene) ที่ทำหน้าที่เป็น transcription factor ควบคุมการสร้างอวัยวะในช่วงตัวอ่อน และควบคุม transcription factor ต่างๆ ความผิดปกติของ PAX6 ทำให้เกิดความผิดปกติแต่กำเนิดต่างๆ ทั่วทั้งลูกตา (เช่น ภาวะไม่มีม่านตา , Peters anomaly, ภาวะจอประสาทตา ดำบกพร่อง)
การกลายพันธุ์ของยีนส่วนใหญ่เป็นชนิด premature truncated codon (PTC) เช่น nonsense และ frameshift และยังมีการรายงานการกลายพันธุ์แบบ missense ด้วย1) การวิเคราะห์ลำดับดีเอ็นเอใน isolated aniridia พบการกลายพันธุ์ของ PAX6 ประมาณ 85%2)
ยีน PAX6 อยู่ติดกับยีน WT1 ซึ่งเป็นยีนต้านมะเร็งบนโครโมโซม 11p13 ในผู้ป่วยประปราย การขาดหายของยีนที่อยู่ติดกันอาจทำให้เกิด WAGR syndrome ซึ่งประกอบด้วย Wilms tumor, aniridia, ความผิดปกติของระบบสืบพันธุ์และทางเดินปัสสาวะ และภาวะปัญญาอ่อน (mental retardation)3) ประมาณ 30% ของผู้ป่วยประปรายจะเกิด Wilms tumor ในระยะแรกทั้งสองข้างก่อนอายุ 5 ปี
หากตรวจพบการกลายพันธุ์ของ PAX6 แต่ไม่พบการขาดหายของ WT1 → สามารถสันนิษฐานได้ว่าไม่มีความเป็นไปได้ของ WAGR syndrome2)
การตรวจทางพันธุกรรมประกอบด้วยการหาลำดับดีเอ็นเอร่วมกับการตรวจหาความผิดปกติของโครงสร้างจีโนมด้วย MLPA/CMA 2)
ในกรณีที่สงสัย WAGR syndrome ในผู้ป่วยที่ไม่มีประวัติครอบครัว แนะนำให้ตรวจทางพันธุกรรม 2)
Q
ควรตรวจยีนของภาวะไม่มีม่านตาหรือไม่?
A
การตรวจยีน PAX6 จำเป็นสำหรับการวินิจฉัยที่แน่ชัด โดยเฉพาะในผู้ป่วยที่ไม่มีประวัติครอบครัว แนะนำให้ตรวจทางพันธุกรรมของ PAX6 และ WT1 เพื่อประเมินความเสี่ยงของ Wilms tumor การตรวจควรทำโดยการหาลำดับดีเอ็นเอร่วมกับ MLPA/CMA ภายใต้การให้คำปรึกษาทางพันธุกรรม ที่เหมาะสม
เกณฑ์การวินิจฉัยและการจำแนกประเภทตามระดับความรุนแรงของโรคต้อหินแต่กำเนิด ชนิดไม่มีม่านตา 1) แสดงไว้ด้านล่าง
ประเภทการวินิจฉัย การรวมกันของเกณฑ์การวินิจฉัย Definite เข้าเกณฑ์ข้อ A ข้อใดข้อหนึ่ง + B1 + E และไม่เข้าเกณฑ์ C น่าจะเป็น (1) เข้าเกณฑ์ข้อ A ใดข้อหนึ่ง + B1 + F และไม่เข้าเกณฑ์ C น่าจะเป็น (2) เข้าเกณฑ์ข้อ A ใดข้อหนึ่ง + B1 + B2 และไม่เข้าเกณฑ์ C น่าจะเป็น (3) เข้าเกณฑ์ข้อ A ใดข้อหนึ่ง + B1 + B3 และไม่เข้าเกณฑ์ C เป็นไปได้ เข้าเกณฑ์ข้อใดข้อหนึ่งของ A + B1 แต่ไม่สามารถแยก C ออกได้อย่างสมบูรณ์
A. อาการ
ความบกพร่องทางการมองเห็น ทั้งสองข้าง (สายตาเสื่อมจากภาวะจอประสาทตา ส่วนกลางเจริญผิดปกติ ต้อกระจก ต้อหิน หรือภาวะกระจกตา เสื่อมจากลิมบัส )
อาการกลัวแสง (ขึ้นอยู่กับระดับของภาวะม่านตา ขาด)
B. ผลการตรวจทางห้องปฏิบัติการ
การตรวจด้วยกล้องจุลทรรศน์ชีวภาพ (slit-lamp) พบความผิดปกติของม่านตา ตั้งแต่การฝ่อบางส่วนจนถึงการไม่มีม่านตา โดยสมบูรณ์ (60-90% เป็นทั้งสองข้าง)
การตรวจอวัยวะภายในลูกตา (fundus) และการตรวจด้วย OCT พบภาวะจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ (foveal hypoplasia) โดยรอยบุ๋มจอตา (foveal pit), เม็ดสีจอตา (macular pigment), และบริเวณไร้หลอดเลือดที่รอยบุ๋มจอตา (foveal avascular zone) ไม่ชัดเจน
การตรวจด้วยกล้องจุลทรรศน์ชีวภาพ (slit-lamp) พบรอยโรคที่กระจกตา เช่น ภาวะขอบกระจกตา เสื่อม (limbal stem cell deficiency) หรือกระจกตา ขุ่น
การตรวจด้วยกล้องจุลทรรศน์ชีวภาพ (slit-lamp) พบต้อกระจก (พบร่วมประมาณ 80%)
ภาวะลูกตาขนาดเล็กจากการตรวจอัลตราซาวนด์ MRI และ CT
อาตา (Nystagmus)ต้อหิน จากการตรวจวัดความดันลูกตา (พบร่วม 50-75%)
C. การวินิจฉัยแยกโรค (โรคที่ต้องแยกออก)
การฝ่อของม่านตา จากการติดเชื้อไวรัสในวงศ์ Herpesviridae ในอดีต
การขาดหายของม่านตา หลังการบาดเจ็บหรือการผ่าตัดภายในลูกตา
คอลโลโบมาของม่านตา ที่เกิดจากการปิดของรอยแยกของถ้วยตาไม่สมบูรณ์
ความผิดปกติของรีเกอร์
กลุ่มอาการไอริสคอร์เนียลเอนโดทีเลียม (ICE)
D. ภาวะแทรกซ้อนนอกตาที่เกี่ยวข้องกับการกลายพันธุ์ของยีน PAX6 (เช่น การขาดหายของคอร์ปัสคัลโลซัม โรคลมชัก)
E. การกลายพันธุ์ทางพยาธิวิทยาของยีน PAX6 หรือการขาดหายของบริเวณ 11p13 (การตรวจทางพันธุศาสตร์)
F. การเกิดโรคในครอบครัว (การถ่ายทอดทางพันธุกรรมแบบออโตโซมัลเด่น 2/3)
การตรวจ วัตถุประสงค์/เนื้อหา การตรวจด้วยกล้องจุลทรรศน์ชีวภาพชนิดร่องกรีด การประเมินระดับความผิดปกติของม่านตา (พื้นฐานการวินิจฉัย) การตรวจอวัยวะภายในลูกตาและ OCT การประเมินภาวะจอประสาทตา ส่วนกลางเจริญไม่เต็มที่ (การหายไปของรอยบุ๋มจอตา และความไม่ชัดเจนของเม็ดสีจอตา) การตรวจมุมลูกตาด้วยกล้อง Gonioscope การประเมินมุมลูกตาผิดปกติและการยึดติดระหว่างรากม่านตา ที่เหลือกับ trabecular meshwork การวัดความดันลูกตา (เป็นประจำ)การตรวจคัดกรองต้อหิน ควรทำเป็นประจำตั้งแต่วัยรุ่นการตรวจอัลตราซาวด์ช่องท้อง การตรวจคัดกรองเนื้องอกวิลมส์ (ในรายที่เกิดประปราย ทุกสองสามเดือน โดยเฉพาะจนถึงอายุ 5 ปี) การตรวจทางพันธุกรรม การระบุการกลายพันธุ์ของยีน PAX6 หรือการขาดหายของบริเวณ 11p13 (จำเป็นสำหรับการวินิจฉัยที่แน่นอน)
ในเด็ก อาจจำเป็นต้องตรวจภายใต้การดมยาสลบ
ในผู้ป่วยที่เกิดเป็นครั้งแรก (sporadic) ประมาณ 30% มีภาวะ Wilms tumor ร่วมด้วย เนื่องจากมีแนวโน้มที่จะเกิดในระยะแรกทั้งสองข้างก่อนอายุ 5 ปี จึงจำเป็นต้องตรวจอัลตราซาวนด์ช่องท้องเป็นระยะทุกสองสามเดือน แนะนำให้ติดตามต่อเนื่องแม้หลังจากอายุ 5 ปี
Q
การวินิจฉัยภาวะไม่มีม่านตา (aniridia) ทำได้อย่างไร?
A
พื้นฐานคือการตรวจหาความผิดปกติของม่านตา ด้วยกล้องจุลทรรศน์ชนิด slit lamp และประเมินภาวะ macular hypoplasia ด้วย OCT การตรวจยีน PAX6 สามารถให้การวินิจฉัยที่แน่ชัด (Definite diagnosis) และในผู้ป่วยที่เกิดเป็นครั้งแรก ควรตรวจหายีน WT1 ร่วมด้วย สิ่งสำคัญคือการแยกจากภาวะม่านตา ฝ่อจากเริม (herpetic iris atrophy), ม่านตา ขาดหลังการบาดเจ็บ, iris coloboma , Rieger anomaly, และ ICE syndrome
ภาวะดวงตาผิดปกติ เช่น ม่านตา ผิดรูป จุดรับภาพชัดเจริญไม่เต็มที่ ตาเล็ก และตากระตุก ยังไม่สามารถรักษาได้ในปัจจุบัน การดูแลหลักคือการติดตามอาการ ส่วนที่ต้องรักษาได้แก่ โรคกระจกตา ต้อกระจก ต้อหิน อาการกลัวแสง และการมองเห็น เลือนราง2)
ในภาวะไม่มีม่านตา จะต้องจัดการแยกกันระหว่างโรคกระจกตา ต้อกระจก ต้อหิน การมองเห็น เลือนราง และอาการกลัวแสง 2)
พื้นที่การรักษา แนวทางการรักษา ความขุ่นของเนื้อกระจกตา การปลูกถ่ายกระจกตา มีข้อจำกัดในการฟื้นฟูการมองเห็น ควรพิจารณาข้อบ่งชี้อย่างรอบคอบภาวะพร่องเซลล์ต้นกำเนิดเยื่อบุกระจกตา พิจารณาการผ่าตัดสร้างพื้นผิวตาใหม่ ต้อกระจก พิจารณาการผ่าตัดตามระดับความขุ่นและอาการกลัวแสง ความดันลูกตา สูง/ต้อหิน รักษาอย่างจริงจังเพื่อรักษาการมองเห็น การดูแลผู้มีสายตาเลือนราง เริ่มตั้งแต่ระยะแรก อาการกลัวแสง จัดการด้วยแว่นตากันแสงหรือคอนแทคเลนส์
ความขุ่นของชั้นเนื้อกระจกตา : การผ่าตัดปลูกถ่ายกระจกตา ช่วยให้การมองเห็น ดีขึ้นได้จำกัดเนื่องจากภาวะแทรกซ้อนของโรคไม่มีม่านตา 2) ในระยะยาว การพยากรณ์โรคทางสายตามักไม่ดีเนื่องจากการเสื่อมของต้อหิน และความล้มเหลวของ graft ที่เกิดขึ้นตามอายุ การปลูกถ่ายกระจกตา ทั้งชั้นเพื่อรักษาความขุ่นของกระจกตา มักไม่ช่วยให้การมองเห็น ดีขึ้น และต้องระวังอัตราการปฏิเสธ graft ที่สูง ในกรณีรุนแรง ควรพิจารณาความสมดุลระหว่างประโยชน์และโทษก่อนดำเนินการ
ภาวะเซลล์ต้นกำเนิดเยื่อบุกระจกตา หมด (LSCD ) : ควรพิจารณาการผ่าตัดรักษา2) โดยเฉพาะการปลูกถ่าย limbal จากผู้อื่น (KLAL ) หรือการปลูกถ่ายเยื่อบุช่องปากที่เพาะเลี้ยง (COMET) สามารถช่วยฟื้นฟูผิวตาได้ในระดับหนึ่ง3) หากมีความขุ่นของชั้นเนื้อกระจกตา ร่วมด้วย การปลูกถ่ายกระจกตา ร่วมด้วยมักมีประโยชน์ในการเพิ่มการมองเห็น 2)
ผู้ป่วยร้อยละ 50–85 จะเกิดต้อกระจก ก่อนอายุ 20 ปี และการผ่าตัดต้อกระจก จะถูกวางแผนตามความรุนแรงของความขุ่นและอาการกลัวแสง 2)
ความเปราะบางของแคปซูลเลนส์และโซนูลาร์ของ Zinn ทำให้การผ่าตัดมีความยากสูง
ต้องระวังความเสี่ยงของโรคต้อหิน ที่แย่ลงหลังผ่าตัด, anterior fibrosis syndrome, และ corneal bullous keratopathy2)
การใส่เลนส์แก้วตาเทียม (IOL ) ต้องพิจารณาอย่างรอบคอบ3)
การใส่ไอริสเทียมร่วมกับการผ่าตัดต้อกระจก อาจทำให้เกิดโรคต้อหิน จึงไม่แนะนำ
ดำเนินการหลังจากให้คำอธิบายอย่างเพียงพอเกี่ยวกับความเสี่ยงที่เกี่ยวข้องกับการผ่าตัด
โรคต้อหิน เกี่ยวข้องโดยตรงกับการพยากรณ์การมองเห็น จึงต้องรักษาอย่างจริงจัง2) โดยใช้แนวทางแบบเป็นขั้นตอนดังนี้
การรักษาด้วยยา : ลดความดันลูกตา ด้วยยาหยอดตาหรือยารับประทาน โดยคำนึงถึงผลข้างเคียงและผลกระทบต่อร่างกายในเด็กการผ่าตัดสร้างทางระบายน้ำออกใหม่ : การผ่าตัดเปิดมุมตาและการผ่าตัดเปิด trabecular meshwork (พิจารณาเมื่อการรักษาด้วยยาไม่ได้ผล)การผ่าตัดกรอง น้ำการผ่าตัดปลูกถ่ายอุปกรณ์ต้อหิน : การผ่าตัดใส่ท่อระบายน้ำชนิดยาว (ต้องได้รับการรับรองจากสถานพยาบาล)การจี้ทำลาย ciliary body : ทางเลือกสุดท้ายเมื่อการรักษาอื่นไม่ได้ผล
การดื้อต่อการรักษาด้วยยาเป็นเรื่องปกติ และการผ่าตัดใส่ท่อระบายน้ำอาจเป็นทางเลือกที่ดี4) เนื่องจากต้อหิน ทำให้สูญเสียลานสายตาแบบถาวร การควบคุมความดันลูกตา ตั้งแต่เนิ่นๆ จึงเป็นกุญแจสำคัญในการรักษาการมองเห็น
Q
โรคต้อหินในภาวะไม่มีม่านตารักษาอย่างไร?
A
เริ่มต้นด้วยการรักษาด้วยยาแบบหยอดหรือรับประทาน แต่มักดื้อต่อยา หากไม่ได้ผลให้พิจารณาผ่าตัดสร้างทางระบายน้ำใหม่ (goniotomy หรือ trabeculotomy) จากนั้นอาจไปสู่การผ่าตัด trabeculectomy หรือการผ่าตัดใส่ท่อระบายน้ำ (glaucoma implant surgery) การผ่าตัดใส่ท่อระบายน้ำต้องได้รับการรับรองจากสถานพยาบาล การจี้ทำลายเลนส์ปรับเลนส์ (cyclophotocoagulation) เป็นทางเลือกสุดท้ายเมื่อการรักษาอื่นไม่ได้ผล จำเป็นต้องติดตามความดันลูกตา อย่างสม่ำเสมอ
การดูแลผู้มีสายตาเลือนราง และการจัดการกับอาการกลัวแสง ควรเริ่มตั้งแต่ระยะแรกเพื่อรักษาการมองเห็น และคุณภาพชีวิต2)
การแก้ไขค่าสายตา : แก้ไขค่าสายตาผิดปกติด้วยแว่นตาเพื่อส่งเสริมพัฒนาการทางการมองเห็น ให้มากที่สุด (พื้นฐาน)แว่นตากันแสง : มีประสิทธิภาพในการลดอาการกลัวแสง ควรจ่ายในกรณีที่มีอาการกลัวแสง รุนแรงSCL ที่มีม่านตาเทียม : มีประโยชน์ทั้งในด้านการปรับปรุงอาการกลัวแสง และรูปลักษณ์ใช้อุปกรณ์ช่วยการมองเห็น เช่น แว่นขยาย แว่นตาสำหรับสายตาเลือนราง หรือเครื่องขยายหนังสือ
ผู้ป่วยภาวะไม่มีม่านตา ส่วนใหญ่สามารถเรียนในโรงเรียนปกติได้ แต่อาจต้องการการสนับสนุนทางการเรียนรู้ เช่น หนังสือเรียนขยายใหญ่ สามารถใช้บริการเรียนบางส่วนในชั้นเรียนสำหรับผู้บกพร่องทางการเห็น หรือขอคำปรึกษาด้านการศึกษาและการเลี้ยงดูจากโรงเรียนคนตาบอดหรือโรงเรียนเฉพาะทางด้านการเห็น นอกจากนี้ยังสามารถยื่นขอรับการช่วยเหลือค่าใช้จ่ายทางการแพทย์ในฐานะโรคหายากที่กำหนดได้ ดังนั้นโปรดปรึกษาแพทย์ผู้ดูแลหรือเจ้าหน้าที่ให้คำปรึกษาทางการแพทย์
ยีน PAX6 เป็นยีนควบคุมหลักที่ถอดรหัสเป็นปัจจัยการถอดรหัสซึ่งควบคุมการสร้างอวัยวะในระยะตัวอ่อน ยีนนี้เริ่มแสดงออกตั้งแต่ระยะแรกของการพัฒนาดวงตาและควบคุมปัจจัยการถอดรหัสต่างๆ การสูญเสียการทำงานของอัลลีลหนึ่งของ PAX6 (haploinsufficiency) ทำให้เกิดความผิดปกติแต่กำเนิดทั่วทั้งดวงตา เช่น aniridia, Peters anomaly, และ macular hypoplasia
การกลายพันธุ์ของ PAX6 ส่วนใหญ่เป็นชนิด PTC เช่น nonsense และ frameshift และยังพบการกลายพันธุ์แบบ missense ด้วย1) การศึกษาความสัมพันธ์ระหว่างจีโนไทป์และฟีโนไทป์ (genotype-phenotype correlation) แสดงให้เห็นว่าความรุนแรงของอาการทางตาขึ้นอยู่กับชนิดของการกลายพันธุ์3)
นอกจากดวงตาแล้ว PAX6 ยังแสดงออกในระบบประสาทส่วนกลาง, เซลล์เกาะแลงเกอร์ฮานส์ของตับอ่อน, และเยื่อบุรับกลิ่น ซึ่งอาจทำให้เกิดภาวะแทรกซ้อนนอกดวงตา เช่น agenesis of corpus callosum, โรคลมชัก, anosmia, และภาวะดื้อต่อกลูโคส เนื่องจากการเจริญผิดปกติของเนื้อเยื่อเหล่านี้1)
มีสองเส้นทางที่คิดว่าเป็นกลไกการเกิดโรคต้อหิน ที่เกี่ยวข้องกับภาวะไม่มีม่านตา
ภาวะมุมเปิด : ความต้านทานการไหลออกของอารมณ์น้ำ ใน trabecular meshwork เพิ่มขึ้นภาวะมุมปิด : รากม่านตา ที่เหลืออยู่บริเวณส่วนปลายสุดยึดติดกับ trabecular meshwork ทำให้เกิดภาวะต้อหินมุมปิด ชนิดหนึ่ง
การเกิดต้อหิน ในวัยทารกพบได้น้อย โดยมักเกิดแบบค่อยเป็นค่อยไปในวัยรุ่นเมื่อโตขึ้น อาจเกิดจากความผิดปกติของมุมลูกตาที่ทำให้มุมเปิด หรือเกิดจากมุมปิดที่ทำให้เกิดต้อหิน
ทางพยาธิวิทยาพบความผิดปกติในการทำงานของเซลล์ต้นกำเนิดเยื่อบุกระจกตา ทำให้เกิดความผิดปกติของเยื่อบุผิวและเยื่อโบว์แมน และเกิดพานนัสที่มีหลอดเลือดมาก การสร้าง Palisades of Vogt ที่บกพร่องนำไปสู่การบุกรุกของเนื้อเยื่อเยื่อบุตา ขาวและการกลายเป็นเคราติน1)
กระจกตา ในภาวะไม่มีม่านตา (aniridia) จะหนากว่าคนปกติ ในวัยเด็กกระจกตา มักปกติ แต่เมื่อโตขึ้นจะเกิดความขุ่นของเนื้อกระจกตา และ LSCD ร่วมด้วย ซึ่งเป็นสาเหตุของการมองเห็น ลดลง การศึกษาในสถาบันเดียวเป็นเวลา 14 ปี (738 ตา) พบว่า aniridia เป็นสาเหตุของ LSCD มากที่สุดถึง 30.9%6)
การพยากรณ์โรคทางสายตาโดยรวมไม่ดี มักอยู่ที่ประมาณ 0.1
ภาวะจอประสาทตา ส่วนมาเคียวเจริญผิดปกติไม่มีวิธีการรักษาที่ได้ผลและเป็นปัจจัยจำกัดการมองเห็น สูงสุด
ความเสียหายของลานสายตาจากโรคต้อหิน เป็นแบบไม่สามารถกลับคืนได้ การควบคุมความดันลูกตา ตั้งแต่เนิ่นๆ จึงมีความสำคัญ
ในกรณีที่เกิดขึ้นโดยไม่มีประวัติครอบครัว ต้องระวังการเกิดเนื้องอกวิลมส์ก่อนอายุ 5 ปี และควรตรวจอัลตราซาวนด์ช่องท้องเป็นประจำ
การศึกษาเกี่ยวกับการพยากรณ์โรคในระยะยาวรายงานว่า การพยากรณ์การมองเห็น โดยทั่วไปไม่ดี แต่มีความแตกต่างกันในแต่ละบุคคลขึ้นอยู่กับชนิดและความรุนแรงของภาวะแทรกซ้อน5)
เนื้อหาต่อไปนี้รวมถึงการรักษาที่ยังอยู่ในขั้นตอนการวิจัยหรือดำเนินการเฉพาะในบางสถานพยาบาลเท่านั้น ไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป การตัดสินใจเกี่ยวกับแนวทางการรักษาควรขึ้นอยู่กับดุลยพินิจของจักษุแพทย์ผู้เชี่ยวชาญเสมอ
ด้วยการแพร่หลายของเทคโนโลยีการหาลำดับรุ่นถัดไป (NGS) อัตราการตรวจพบการกลายพันธุ์ของ PAX6 ใน isolated aniridia อยู่ที่ประมาณ 85% 2) การตรวจด้วยไมโครอาร์เรย์โครโมโซม (CMA) มีความไวสูงกว่าการตรวจโครโมโซมแบบดั้งเดิมในการตรวจหาการขาดหายของชิ้นส่วนเล็กๆ ที่ 11p13 และมีส่วนช่วยในการเพิ่มความแม่นยำในการวินิจฉัย WAGR syndrome 2)
มีการสะสมข้อมูลผลลัพธ์ระยะยาวของการปลูกถ่ายเยื่อบุช่องปากที่เพาะเลี้ยง (COMET) มากขึ้น2) สำหรับ Boston type I keratoprosthesis มีรายงานว่าการมองเห็น ดีขึ้น 65–93% ในระยะสั้น (17–28.7 เดือน) แต่ลดลงเหลือ 43.5% ที่ 4.5 ปี2)
HumanOptics CustomFlex ArtificialIris เป็นอุปกรณ์ม่านตาเทียม ซิลิโคนที่สั่งทำเฉพาะบุคคล ซึ่งมีประโยชน์ในการลดอาการกลัวแสง และปรับปรุงลักษณะรูปลักษณ์ภายนอก แต่ยังไม่ได้รับการอนุมัติในญี่ปุ่น ณ ปี 2024 การรักษาแบบกำหนดเป้าหมายระดับโมเลกุลที่มุ่งเป้าไปที่ PAX6 haploinsufficiency ยังอยู่ในขั้นตอนการวิจัยและยังไม่ถึงการประยุกต์ใช้ทางคลินิก3)
大家義則, 川崎諭, 西田希, 木下茂, 外園千恵, 大橋裕一, 他. 無虹彩症の診断基準および重症度分類. 日眼会誌. 2020;124:83-88.
厚生労働科学研究費補助金難治性疾患政策研究事業「角膜難病の標準的診断法および治療法の確立を目指した調査研究」研究班. 無虹彩症の診療ガイドライン. 日眼会誌. 2021;125:38-73.
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
American Academy of Ophthalmology. Diagnosis and Management of Aniridia. EyeNet Magazine. 2014. https://www.aao.org/eyenet/article/diagnosis-management-of-aniridia
Japanese Ophthalmological Society. Clinical practice guideline for aniridia. Jpn J Ophthalmol. 2026. doi:10.1007/s10384-025-01296-y. https://link.springer.com/article/10.1007/s10384-025-01296-y
Hu JCW, Weissbart SB. Limbal stem cell deficiency and severe ocular surface disease: a review. Ann Eye Sci. 2023;8:35.