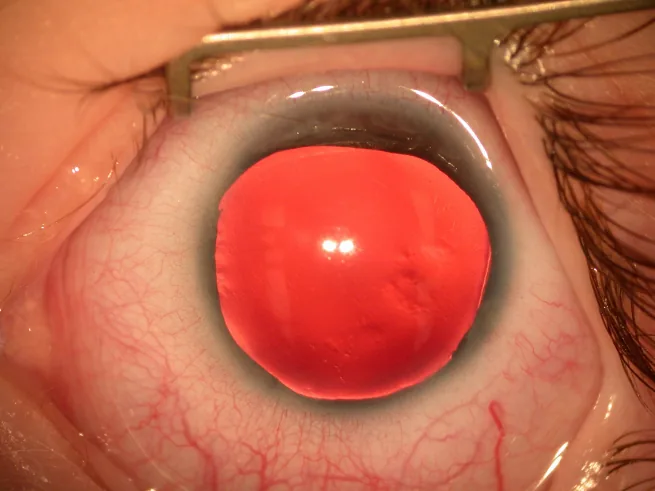
Aniridie ist ein angeborener Zustand, bei dem die Iris vollständig oder unvollständig fehlt. Obwohl sie als „Aniridie“ bezeichnet wird, bleibt oft ein Irisrest im äußersten Kammerwinkelbereich erhalten.
Im Jahr 2017 wurde sie als designierte seltene Krankheit nach dem japanischen Gesetz über seltene Krankheiten anerkannt 1). Patienten, bei denen die Krankheit diagnostiziert wird und die einen Schweregrad von III oder höher aufweisen, haben Anspruch auf Kostenübernahme für medizinische Behandlungen, mit einer einkommensabhängigen Obergrenze für die Eigenbeteiligung 2).
Epidemiologische Studien aus Schweden und Norwegen berichten eine Prävalenz von etwa 1 zu 90.000 Personen3). Eine detaillierte ophthalmologische Bewertung von 43 Fällen mit PAX6-Genmutationen zeigte, dass das Ausmaß der Irisdysplasie je nach Mutationstyp variiert3).
QIst Aniridie erblich?
A
Etwa zwei Drittel der Fälle sind autosomal-dominant vererbt, mit einer 50%igen Wahrscheinlichkeit der Vererbung von einem betroffenen Elternteil auf das Kind. Das restliche Drittel tritt sporadisch ohne Familienanamnese auf. Bei sporadischen Fällen besteht ein Risiko für das WAGR-Syndrom, das mit Wilms-Tumor (Nierentumor) einhergeht, daher wird eine genetische Untersuchung der PAX6- und WT1-Gene empfohlen.
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Spaltlampenfoto des vorderen Augenabschnitts: Die Iris ist nahezu vollständig fehlend, nur am Rand ist ein sehr dünner Irisrest sichtbar. Dies zeigt direkt den typischen klinischen Befund der Aniridie und eignet sich zur Veranschaulichung der Hauptsymptome und klinischen Befunde.
Aufgrund des fehlenden oder unvollständigen Irisgewebes funktioniert die Pupille nicht, sodass die in das Auge einfallende Lichtmenge nicht reguliert werden kann. Daher klagen die Patienten über starke Photophobie. Zudem führt die foveale Hypoplasie zu einer schlechten Fixation, was häufig ab dem frühen Säuglingsalter das Hauptsymptom eines horizontalen Nystagmus ist.
Photophobie: Unfähigkeit der Iris, die Lichtmenge zu regulieren → starke Blendempfindlichkeit
Nystagmus (horizontaler Nystagmus): Schlechte Fixation aufgrund fovealer Hypoplasie. Tritt ab dem frühen Säuglingsalter auf.
Irisdysplasie: unterschiedliche Ausprägungen von partieller Atrophie bis hin zu vollständigem Fehlen
Foveale Hypoplasie: nahezu in allen Fällen vorhanden. Fehlende Fovea-Einsenkung und unklare Makulapigmentierung. Hauptfaktor für die Sehschärfenminderung.
Schielen (Strabismus): Tritt aufgrund der schlechten Sehschärfe auf.
Erworbene Komplikationen (treten mit dem Wachstum auf)
Grauer Star (Katarakt): Tritt bei etwa 80% auf. Bis zum 20. Lebensjahr entwickeln 50–85% eine Katarakt.
Grüner Star (Glaukom): Tritt bei 50–75% auf. Im Säuglingsalter selten, entwickelt sich fortschreitend im Jugendalter.
Limbusstammzellinsuffizienz (LSCD): Im frühen Kindesalter meist normal, aber mit zunehmendem Alter fortschreitende Hornhauttrübung und Vaskularisation.
Zusammenfassung der Häufigkeit von Augenkomplikationen
Das PAX6-Gen wird nicht nur im Augengewebe, sondern auch im Zentralnervensystem, in den Langerhans-Inseln der Bauchspeicheldrüse und im Riechepithel exprimiert. Eine Hypoplasie dieser Gewebe kann zu verschiedenen extraokulären Komplikationen führen1).
Die Sehprognose ist im Allgemeinen schlecht und liegt meist bei etwa 0,1. Je nach Ausmaß der fovealen Hypoplasie und Vorhandensein von Komplikationen kann sie jedoch zwischen 0,1 und 0,7 variieren. Für die foveale Hypoplasie gibt es derzeit keine wirksame Behandlung; sie ist der größte limitierende Faktor für das Sehvermögen. Durch geeignete Refraktionskorrektur und Low-Vision-Versorgung kann die Lebensqualität verbessert werden.
Die Ursache der Aniridie ist der Funktionsverlust eines Allels (Haploinsuffizienz) des PAX6-Gens auf dem kurzen Arm von Chromosom 11 (11p13). Sie entsteht durch die Halbierung der funktionellen Gendosis. Bei einer Anomalie beider Allele wird angenommen, dass dies embryonal letal ist1).
PAX6 ist ein Master-Kontrollgen für Transkriptionsfaktoren, die die Organentwicklung in der Embryonalphase steuern, und reguliert verschiedene Transkriptionsfaktoren. Eine Anomalie von PAX6 führt zu verschiedenen angeborenen Anomalien des gesamten Auges (Aniridie, Peters-Anomalie, foveale Hypoplasie usw.).
Die Art der Genmutationen sind häufig Nonsense- oder Frameshift-Mutationen, die zu einem vorzeitigen Abbruchkodon (PTC) führen, aber auch Missense-Mutationen wurden berichtet1). Bei der Sequenzierungsanalyse der isolierten Aniridie werden in etwa 85% der Fälle PAX6-Mutationen nachgewiesen2).
Das PAX6-Gen liegt auf Chromosom 11p13 in unmittelbarer Nähe zum Tumorsuppressorgen WT1. Bei sporadischen Fällen kann eine Deletion benachbarter Gene zum WAGR-Syndrom führen, das aus Wilms-Tumor, Aniridie, urogenitalen Anomalien und geistiger Retardierung besteht3). Bei etwa 30% der sporadischen Fälle tritt ein Wilms-Tumor vor dem 5. Lebensjahr frühzeitig beidseitig auf.
PAX6-Mutation positiv, keine WT1-Deletion → WAGR-Syndrom ist wahrscheinlich ausgeschlossen2)
Die genetische Testung erfolgt durch eine Kombination aus DNA-Sequenzierung und MLPA/CMA zum Nachweis genomischer Strukturanomalien2)
Bei sporadischen Fällen mit Verdacht auf WAGR-Syndrom wird eine genetische Untersuchung empfohlen 2)
QSollte ich einen Gentest für Aniridie durchführen lassen?
A
Ein PAX6-Gentest ist zur Bestätigung einer Definitiv-Diagnose erforderlich, insbesondere bei sporadischen Fällen wird eine genetische Untersuchung von PAX6 und WT1 zur Bewertung des Wilms-Tumor-Risikos empfohlen. Die Untersuchung sollte DNA-Sequenzierung mit MLPA/CMA kombinieren und unter angemessener genetischer Beratung durchgeführt werden.
Wilms-Tumor-Screening (sporadische Fälle, alle paar Monate, besonders bis zum 5. Lebensjahr)
Gentest
Identifizierung einer PAX6-Genmutation oder einer Deletion der Region 11p13 (für definitive Diagnose erforderlich)
Bei Kindern kann eine Untersuchung unter Vollnarkose erforderlich sein.
QWie wird die Diagnose einer Aniridie gestellt?
A
Grundlage ist der Nachweis einer Irisdysplasie mittels Spaltlampenmikroskopie und die Beurteilung einer fovealen Hypoplasie mittels OCT. Eine definitive Diagnose ist durch PAX6-Gentest möglich; bei sporadischen Fällen wird auch eine WT1-Gensuche durchgeführt. Die Abgrenzung zu Herpes-Irisatrophie, posttraumatischem Irisdefekt, Iriskolobom, Rieger-Anomalie und ICE-Syndrom ist wichtig.
Irisdysplasie, foveale Hypoplasie, Mikrophthalmus und Nystagmus sind derzeit nicht behandelbar; die Grundlage ist die Beobachtung. Behandlungsziele sind Keratopathie, Katarakt, Glaukom, Photophobie und Sehbehinderung2).
Hornhauttrübung: Die Verbesserung der Sehfunktion durch Hornhauttransplantation ist aufgrund von Komplikationen der Aniridie begrenzt2). Langfristig ist die Sehprognose aufgrund von Glaukomverschlechterung und allmählichem Transplantatversagen oft schlecht. Eine perforierende Keratoplastik bei Hornhauttrübung führt oft nicht zu einer Sehverbesserung, und die Abstoßungsrate ist hoch. Bei schweren Fällen sollte der Eingriff nach sorgfältiger Abwägung von Nutzen und Risiken durchgeführt werden.
Limbale Stammzellinsuffizienz (LSCD): Eine chirurgische Behandlung sollte in Betracht gezogen werden2). Konkret kann durch allogene Limbustransplantation (KLAL) oder kultivierte orale Mukosaepitheltransplantation (COMET) eine gewisse Rekonstruktion der Augenoberfläche erreicht werden3). Bei gleichzeitiger Hornhauttrübung ist eine kombinierte Hornhauttransplantation oft zur Verbesserung der Sehkraft nützlich2).
Bis zum Alter von 20 Jahren entwickeln 50–85 % einen Katarakt, und die Kataraktoperation wird je nach Schweregrad der Trübung und Photophobie geplant2).
Hoher operationsbedingter Schwierigkeitsgrad aufgrund der Fragilität der Linsenkapsel und der Zonulafasern
Risiko einer postoperativen Glaukomverschlechterung, eines anterioren Fibrosesyndroms und einer bullösen Keratopathie beachten2)
Die Implantation einer Intraokularlinse (IOL) ist eine sorgfältig abzuwägende Indikation3)
Die gleichzeitige Implantation einer künstlichen Iris während der Kataraktoperation wird nicht empfohlen, da sie ein Glaukom auslösen kann
Der Eingriff wird nach ausführlicher Aufklärung über die operationsbedingten Risiken durchgeführt.
Da das Glaukom die Sehprognose direkt beeinflusst, wird es aktiv behandelt2). Es wird ein schrittweiser Ansatz verfolgt:
Medikamentöse Therapie: Augeninnendrucksenkung durch Augentropfen und orale Medikamente unter Berücksichtigung von Nebenwirkungen und systemischen Auswirkungen bei Kindern
Abflusswegsrekonstruktion: Goniotomie oder Trabekulotomie (bei Unwirksamkeit der medikamentösen Therapie in Betracht ziehen)
Zyklophotokoagulation: Letzte Option, wenn andere Behandlungen versagen
Häufig besteht eine Resistenz gegen medikamentöse Therapie, sodass eine Tubus-Shunt-Operation eine gute Option sein kann4). Da Gesichtsfeldausfälle bei Glaukom irreversibel sind, ist eine frühzeitige Augeninnendruckkontrolle der Schlüssel zum Erhalt der Sehfunktion.
QWie wird das Glaukom bei Aniridie behandelt?
A
Zunächst erfolgt eine medikamentöse Therapie mit Augentropfen oder oralen Medikamenten, jedoch spricht sie oft nicht an. Bei unzureichender Wirkung wird eine Wiederherstellung des Abflusstrakts (Goniotomie, Trabekulotomie) in Betracht gezogen, gefolgt von einer Trabekulektomie oder einer Langtubus-Operation (Glaukom-Implantat-Operation). Die Langtubus-Operation erfordert eine Einrichtungszertifizierung. Die Zyklophotokoagulation ist die letzte Option, wenn andere Behandlungen versagen. Eine regelmäßige Augeninnendrucküberwachung ist unerlässlich.
Das PAX6-Gen ist ein Master-Kontrollgen, das einen Transkriptionsfaktor kodiert, der die Organentwicklung in der Embryonalphase steuert. Es wird ab dem frühen Augenbulbus exprimiert und reguliert verschiedene Transkriptionsfaktoren. Der Funktionsverlust eines Allels (Haploinsuffizienz) von PAX6 führt zu angeborenen Anomalien des gesamten Auges (Aniridie, Peters-Anomalie, Makulahypoplasie usw.).
PAX6-Mutationen sind häufig vom PTC-Typ (Nonsense- oder Frameshift-Mutationen), aber auch Missense-Mutationen wurden berichtet1). Studien zur Genotyp-Phänotyp-Korrelation zeigen, dass die Schwere der ophthalmologischen Befunde je nach Mutationstyp variiert3).
PAX6 wird auch außerhalb des Auges im Zentralnervensystem, in den Langerhans-Inseln der Bauchspeicheldrüse und im Riechepithel exprimiert, sodass extraokuläre Komplikationen (Agenesie des Corpus callosum, Epilepsie, Anosmie, Glukoseintoleranz) durch Hypoplasie dieser Gewebe auftreten können1).
Für die Pathogenese des Glaukoms bei Aniridie werden zwei Wege angenommen.
Offenwinkelpathologie: Erhöhter Abflusswiderstand des Kammerwassers im Trabekelwerk
Winkelblockpathologie: Die am äußersten Rand verbliebene Iriswurzel verklebt mit dem Trabekelwerk und führt zu einer Art Winkelblockglaukom
Ein Glaukom im Säuglingsalter ist selten; es entwickelt sich fortschreitend im Jugendalter. Es kann durch eine Kammerwinkeldysgenesie im offenen Zustand oder durch einen Winkelblock entstehen.
Pathologisch zeigt sich eine Funktionsstörung der epithelialen Stammzellen der Hornhaut, die zu Anomalien des Epithels und der Bowman-Membran sowie zur Bildung eines vaskularisierten Pannus führt. Eine Hypoplasie der Palisaden von Vogt führt zum Eindringen von Bindehautgewebe und zur Keratinisierung1).
Die Hornhaut bei Aniridie ist dicker als bei Gesunden. Im Kindesalter ist die Hornhaut oft normal, aber mit zunehmendem Alter treten Hornhautstromatrübung und LSCD auf, die zu Sehverschlechterung führen. In einer 14-jährigen Einzelstudie (738 Augen) war Aniridie mit 30,9 % die häufigste Ursache für LSCD6).
Die Sehprognose ist meist schlecht und liegt oft bei etwa 0,1
Die Makulahypoplasie hat keine wirksame Behandlung und ist der größte limitierende Faktor für das Sehvermögen
Gesichtsfeldausfälle durch Glaukom sind irreversibel, daher ist eine frühzeitige Augeninnendruckkontrolle wichtig
Bei sporadischen Fällen ist auf eine frühe Entwicklung eines Wilms-Tumors vor dem 5. Lebensjahr zu achten, und regelmäßige abdominale Ultraschalluntersuchungen sollten fortgesetzt werden.
Studien zur Langzeitprognose zeigen, dass die Sehprognose im Allgemeinen schlecht ist, jedoch je nach Art und Schwere der Komplikationen individuell variiert 5).
Durch die Verbreitung der Next-Generation-Sequenzierung (NGS) liegt die Nachweisrate von PAX6-Mutationen bei isolierter Aniridie bei etwa 85 % 2). Chromosomale Microarrays (CMA) sind empfindlicher als herkömmliche Chromosomentests für den Nachweis von Mikrodeletionen in 11p13 und tragen zur verbesserten Diagnosegenauigkeit des WAGR-Syndroms bei 2).
Die Langzeitergebnisse der kultivierten oralen Mukosa-Epitheltransplantation (COMET) werden zunehmend dokumentiert 2). Für die Boston-Typ-I-Keratoprothese wird berichtet, dass kurzfristig (17–28,7 Monate) bei 65–93 % eine Sehverbesserung erreicht wird, die jedoch nach 4,5 Jahren auf 43,5 % abfällt 2).
Künstliche Iris-Implantate und Perspektiven der Gentherapie
Die HumanOptics CustomFlex ArtificialIris ist ein individuell angefertigtes künstliches Irisimplantat aus Silikon, das zur Linderung von Photophobie und zur Verbesserung des Aussehens nützlich ist, jedoch in Japan bis 2024 nicht zugelassen ist. Die auf PAX6-Haploinsuffizienz abzielende molekulare Therapie befindet sich derzeit in der Forschungsphase und hat noch keine klinische Anwendung erreicht 3).
Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.